- Muco
-
Mucoviscidose
Mucoviscidose Autre nom Fibrose kystique du pancréas Référence MIM 219700 Transmission Autosomique récessive Chromosome Chromosome 7 7q31.2 Gène CFTR Empreinte parentale Non Mutation Ponctuelle Mutation de novo ? Nombre d'allèles pathologiques Plus de 1 500, la plus fréquente est DeltaF508[1] Anticipation ? Porteur sain environ 1/25 soit 4% de la population générale occidentale[2] Incidence 1 sur 2500 naissances[3] Prévalence 5 à 6 000 cas en France[4] et 70 000 cas dans le monde[5] Pénétrance Plus faible chez les garçons Nombre de cas Maladie génétiquement liée Non Diagnostic prénatal Possible Article principal - Liste des maladies génétiques à gène identifié Mucoviscidose
Classification et ressources externesCIM-10 E84 CIM-9 277.0 La mucoviscidose (pour « maladie des mucus visqueux » en français) [6] ou cystic fibrosis (pour « fibrose kystique », sous-entendue du pancréas, en anglais)[7] est une maladie génétique, affectant les épithéliums glandulaires de nombreux organes. C'est la maladie génétique létale à transmission autosomique récessive la plus fréquente dans les populations de type europoïde, alors qu'elle est très rare dans les populations africaines et asiatiques. Elle est liée à des mutations du gène CFTR sur le chromosome 7, entraînant une altération de la protéine CFTR (sigle pour cystic fibrosis transmembrane conductance regulator). Cette protéine est un canal ionique perméable au chlore dont la fonction est de réguler le transport du chlore à travers les membranes cellulaires. Son dysfonctionnement provoque une augmentation de la viscosité du mucus et son accumulation dans les voies respiratoires et digestives. La maladie touche de nombreux organes mais les atteintes respiratoires sont prédominantes et représentent l'essentiel de la morbidité. La forme clinique la plus fréquente associe troubles respiratoires, troubles digestifs et troubles de la croissance staturopondérale. D'évolution chronique et progressive, la maladie s'exprime souvent tôt dès la petite enfance même s'il existe des formes frustes de diagnostic tardif.
Le diagnostic biologique repose sur le test de la sueur confirmé par une identification des mutations génétiques. Le dépistage néonatal, généralisé en France depuis 2002 permet un diagnostic et une prise en charge précoce alors que le conseil génétique permet à un couple hétérozygote connu de ne pas avoir un autre enfant malade. Il n'y a pas de traitement curatif mais les progrès de la prise en charge ont permis d'améliorer la qualité et l'espérance de vie des patients ; ainsi en France, l'espérance de vie à la naissance[8] est passée de 7 ans en 1965 à 47 ans en 2005[9].
Connue depuis le Moyen Âge, la maladie est décrite scientifiquement par le pédiatre suisse Guido Fanconi en 1936[10]. Elle est identifiée deux ans plus tard par Dorothy Hansine Andersen comme une entité pathologique atteignant le pancréas d'où son nom historique de fibrose kystique du pancréas. Elle conserve ce nom en anglais : cystic fibrosis.
Historique
Grandes dates

Histoire de la description de la maladie
 Dorothy Hansine Andersen fut la première à décrire la mucoviscidose en tant que maladie à part entière.(photo de la National Library of Medicine)
Dorothy Hansine Andersen fut la première à décrire la mucoviscidose en tant que maladie à part entière.(photo de la National Library of Medicine)
La mucoviscidose, décrite scientifiquement comme maladie en 1936, était en fait déjà connue depuis longtemps. Au Moyen Âge on connaissait le sort funeste du nouveau-né dont la mère remarquait le « baiser salé », c'est-à-dire le goût salé laissé par un baiser sur le front de l'enfant[11],[12],[13]. Welsh cite un vieil adage du folklore de l'Europe du Nord[12] : « Malheur à l'enfant chez qui un baiser sur le front a un goût salé. Il est ensorcelé et doit bientôt mourir. ». En 1606, le médecin espagnol Alonso y de los Ruyzes de Fonteca rapporte que les doigts passés sur le front des enfants ensorcelés ont un goût salé[14]. Rochholz dans son almanach de 1857 écrit « L'enfant mourra bientôt dont le front embrassé est salé. » [11]. Selon Busch[13] une des premières descriptions médicales des lésions pancréatiques rencontrées dans la mucoviscidose pourrait être un rapport d'autopsie fait par le professeur Pieter Pauw, à Leyde aux Pays-Bas en 1595, dans lequel il décrit une patiente chétive de 11 ans supposée ensorcelée et présentant un pancréas élargi, dur et blanc.
Au XIXe siècle le médecin viennois Karel Rokitansky signale chez un fœtus un cas fatal de péritonite méconiale[15], qui sera plus tard identifiée comme une complication de l'iléus méconial, obstruction intestinale néonatale présente chez certains enfants mucoviscidosiques décrite en 1905 par Landsteiner[16].
Au début du XXe siècle apparaissent les premières observations associant maladie pulmonaire, diarrhée et anomalie pancréatique avec plusieurs cas dans la même famille. En 1912, Garrod[17] décrit des familles dont les enfants présentent une diarrhée graisseuse et meurent d'infection pulmonaire. Ces descriptions se focalisent le plus souvent sur les problèmes digestifs, stéatorrhée et troubles pancréatiques, et leurs auteurs en font une forme de maladie cœliaque même si les problèmes bronchopulmonaires sont également notés[18],[19],[10],[20].
C'est en 1936, dans une thèse écrite en allemand et présidée par le pédiatre suisse Guido Fanconi, que la maladie est décrite pour la première fois chez des enfants supposés atteints de maladie cœliaque, sous le nom de « fibrose kystique du pancréas et bronchectasie »[10]. La mucoviscidose ne fut considérée comme une entité pathologique distincte qu'en 1938 par la pédiatre américaine Dorothy Hansine Andersen, médecin au Babies' Hospital de New York, qui publia un article intitulé « fibrose kystique du pancréas et ses relations avec la maladie cœliaque » [18]. C'est en pratiquant des autopsies sur des nourrissons qu'elle décrivit les caractéristiques cliniques et histologiques de la maladie, notamment l'obstruction intestinale néonatale, les complications respiratoires et digestives et les lésions histologiques spécifiques du pancréas. Elle relia cette maladie à un déficit en vitamine A[21] et persista à soutenir cette théorie[22],[23] pendant de nombreuses années bien qu'elle ne fût jamais confirmée.
Le terme de mucoviscidosis, créé à partir des termes « mucus » et « visqueux », fut utilisé pour la première fois en 1943 par le docteur Sydney Farber[24], médecin-chef au Children's Hospital de Boston, afin de corriger la dénomination employée par Dorothy Andersen, centrée sur le pancréas. Farber était persuadé que la maladie était due à une diffusion généralisée de mucus visqueux. Le terme de mucoviscidosis reste très employé dans le monde, et notamment en France, et est parfois préféré au terme anglais cystic fibrosis[25].
Le caractère héréditaire et le mode de transmission récessif furent suggérés en 1945 par Dorothy Andersen et Hodges[26].
C'est à la suite d'une vague de chaleur entraînant un état de prostration chez de jeunes patients de l'hôpital Columbia de New York en 1948[27] que le docteur Paul di Sant' Agnese découvre et décrit en 1953 les anomalies électrolytiques[28],[29] dans la sueur des malades (augmentation importante du chlore, du sodium et moins marquée du potassium), permettant d'envisager un diagnostic spécifique à la maladie : le test de la sueur. Jusqu'alors, le diagnostic ne pouvait être fait que devant la combinaison de signes cliniques et de symptômes évocateurs d'insuffisance pancréatique et de malabsorption intestinale. Pendant des décennies la stimulation thermique a été utilisée, au cours de laquelle les enfants étaient entièrement enveloppés dans des pansements afin de stimuler la transpiration. Le recueil de la sueur était effectué grâce à des buvards. De réalisation difficile, la technique fut par la suite simplifiée par la méthode de l'iontophorèse à la pilocarpine décrite en 1959 par Gibson et Cooke[30], puis améliorée par Shwachman[31] et standardisée chez l'enfant par Legrys[32]. Le test de la sueur devint et reste jusqu'à ce jour le test le plus fiable pour établir le diagnostic, en dehors de l'analyse génétique qui ne fut disponible que bien plus tard.
C'est au début des années 1980 que le lien physiopathologique fut fait entre d'une part l'anomalie de la sécrétion de mucus, entraînant des obstructions glandulaires avec anomalies histologiques et d'autre part l'anomalie de la sueur, entraînant des sécrétions salées sans anomalie histologique. En 1981, Knowles et al.[33] découvrirent que le potentiel électrique au niveau de la muqueuse nasale des patients mucoviscidosiques étaient plus électronégatifs que chez les sujets sains, ce qu'ils expliquèrent par une réabsorption massive du sodium entraînant une déshydratation à la surface de l'épithélium. Cette découverte faisait le lien physiologique entre les poumons, le pancréas et les glandes sudoripares. Le lien commun expliquant l'atteinte des différents organes n'était pas le mucus lui-même, mais les anomalies électrolytiques. En 1983 Quinton, lui-même atteint de la mucoviscidose, montra que l'imperméabilité au chlore qu'il avait trouvée dans les glandes sudoripares[34] était la cause de l'augmentation des électrolytes dans la sueur[35]. Ceci fut considéré comme une étape majeure dans la compréhension de la maladie.
Restait à localiser et à identifier le gène dont la mutation provoque la mucoviscidose, une tâche rendue difficile car seules la sémiologie clinique, le mode de transmission autosomique récessif et l'anomalie de transport du chlore étaient alors connus. En l'absence de toute connaissance sur la protéine défectueuse, et donc de marqueur chimique identifié, la récente technique du clonage positionnel - ou génétique inverse, qui permet d'identifier un gène sans connaître la protéine qu'il code, fut utilisée. Cette méthode consiste grâce à des analyses statistiques de liaison génétique à déterminer la région du chromosome où le gène a une forte probabilité de se trouver, afin de la séquencer et d'étudier les gènes exprimés. Le gène de la mucoviscidose est le premier gène à avoir été cloné uniquement par analyse de liaisons[36].
En 1985, Eiberg et al.[37] retrouvent un lien entre l'enzyme paraoxinase (PON) et le gène CF en étudiant des familles comportant plus d'un enfant atteint. La même année, Tsui et al.[38], suite à des études sur des souris hybrides, localisent le gène sur le bras long du chromosome 7 grâce à un marqueur RLFP lié à la fois à la paraoxinase et à la mucoviscidose.
En 1989, le gène impliqué dans la mucoviscidose est isolé par les équipes de Lap-Chi Tsui, Collins et Riordan[39],[40],[41]. L'anomalie génétique à l'origine de la maladie est enfin découverte, il s'agit d'une mutation d'un gène localisé en 7q31 et contenant 27 exons, nommé cystic fibrosis (CF) codant une protéine transmembranaire appelée cystic fibrosis transmenbrane conductance regulator (CFTR) composée de 1480 acides aminés. Ce n'est qu'un peu plus tard qu'on apporta les preuves que CFTR était bien un canal du chlore[42],[43]. La découverte de l'anomalie génétique permit par la suite d'ajouter le génotypage au protocole diagnostique, et d'envisager la thérapie génique.
Histoire de la prise en charge de la maladie
Dans les années 1940, la maladie était considérée comme étant avant tout un problème nutritionnel avec une déficience en vitamine A. La prise en charge se résumait essentiellement à un régime riche en protéines, des injections intramusculaires de vitamine A à hautes doses, des extraits pancréatiques et des inhalations de pénicilline[44]. En 1945, Dorothy Andersen conseille « un régime pauvre en graisse, riche en protéines, avec une proportion libre de légumes, fruits et sucres et une restriction modérée en féculents. Une supplémentation en vitamine A est essentielle et des complexes pancréatiques et de la vitamine B sont ajoutés. » [45]. Elle précise que la diète doit être débutée précocement pour être efficace[23]. À cette époque la durée de survie reste courte, 64% des 28 patients d'une série de la Mayo Clinic n'atteignant pas l'âge de 7 ans[46]. La première drogue antibactérienne, un sulfamidé commercialisé sous le nom de Prontosil, fut disponible en 1934 et la pénicilline sous forme injectable en 1944. D'autres antibiotiques suivirent et eurent un rôle essentiel dans le traitement des patients. À cette date, le principal germe pathogène était Staphylococcus aureus et de nombreuses souches étaient encore sensibles à la pénicilline[44]. En 1946 Di Sant Agnese et D. Andersen écrivent que l'amélioration du pronostic lors de cette décennie est due à « un régime approprié débuté rapidement et poursuivi de façon continue, l'utilisation de sulphadiazine pendant la période de toux chronique et de nébulisation de pénicilline. » [47].
Dans les années 1950, le sort des enfants atteints est toujours considéré comme sans espoir mais quelques centres spécialisés dans la prise en charge de la mucoviscidose voient le jour aux États-Unis et au Royaume-Uni. Le drainage postural est alors un des traitements traditionnels au stade de bronchectasie et en 1950 Young propose de le débuter dès le diagnostic posé[44]. L'effet des bronchodilateurs est décrit en 1959 [48]. En 1955 Shwachman décrit de façon détaillée une méthode de prise en charge qui pose les fondements du traitement moderne : diagnostic précoce, traitement actif et précoce de l'infection pulmonaire et surveillance et maintien de l'état nutritionnel[44]. Lors de cette décennie, d'autres antibiotiques apparaissent. Dès 1951, alors que Staphylococcus aureus est la bactérie habituellement retrouvée, on note l'augmentation de la fréquence de Pseudomonas aeruginosa, attribuée aux traitements antibiotiques prolongés[49], néanmoins le bénéfice de traitements antibiotiques agressifs devient progressivement évident[50]. En 1952 Shwachman rapporte l'apparition de résistance de S. aureus à la terramycine et écrit que les aérosols de pénicilline et de streptomycine sont utiles[51]. En 1958 Shwachman et Kulczycki publient le premier article de synthèse provenant d'un grand centre spécialisé dans lequel ils décrivent leur système de classification clinique et notent une amélioration du pronostic des enfants atteints et une augmentation de la fréquence de survie jusqu'à l'âge adulte[52]. La mortalité des enfants malades avec un ileus méconial est alors toujours supérieure à 50% mais se trouve grandement améliorée par une méthode chirurgicale d'iléostomie décrite en 1957 par Bishop et Koop[53]. C'est en 1955 que la preuve objective de l'efficacité d'un traitement par enzymes pancréatiques est apportée par l'équipe londonienne de Norman[54].
Dans les années 1960, la création d'organisations nationales consacrée à la maladie permet une approche collaborative entre la communauté médicale et les parents et patients. Ainsi la US National CF Research Foundation (qui deviendra plus tard la CF Foundation) est créé en 1955, la Canadian CF Foundation en 1959, la UK CF Research Foundation Trust en 1964 et l'International Cystic Fibrosis Association en 1965 à Paris. La survie des malades reste très précaire, la plupart décédant durant la jeune enfance, l'enfance ou l'adolescence, après de pénibles années de maladie chronique[44]. La « Mist tent therapy » fut une forme de traitement populaire aux USA à cette période qui consistait à faire dormir les enfants sous une fine brume afin de diminuer la viscosité des secrétions et d'améliorer leur état respiratoire[55],[56]. Mais son efficacité ne fut pas reconnue et le traitement fut discrédité au début des années 1970[57], [58], [59]. Des résultats encourageants proviennent néanmoins de Londres, en 1969 David Lawson est le premier à suggérer qu'une antibioprophylaxie anti-staphylococcique continue pourrait améliorer la survie[60] et en 1972 il écrit qu'« un diagnostic plus rapide par un dépistage néonatal amélioré est essentiel car actuellement le diagnostic est posé au stade alors que des lésions pulmonaires sont déjà présentes » [61]. Margaret Means en 1972 également rapporte une réduction de moitié de la mortalité chez les enfants de moins de 5 ans, passant de 14% à 6.5%, grâce à des antibiotiques anti-staphylococciques disponibles depuis 1957. Elle écrit qu'« un traitement vigoureux et un contrôle de l'infection dès la petite enfance, peut prévenir la plupart de ces patients de devenir insuffisants respiratoires à l'enfance » [62].
Dans les années 1970, de nombreux progrès sont réalisés, notamment au niveau des soins chirurgicaux et médicaux néonataux; la ventilation des nouveau-nés devient possible et s'améliore progressivement. Cette décénie est marquée par l'intérêt croissant vis à vis des problèmes nutritionnels chroniques qui apparaissent avec l'extension de la survie des patients[44]. L'attitude générale était alors restrictive et rigide, on recommandait alors des apports de 30 à 40 g de graisses maximum par jour, 200 Kcal/kg/jour et de 4 à 5 g de protéine/kg/jour » [63]. L'« Allan Diet » destinée à améliorer l'absorption intestinale et l'état nutritionnel des patients[64] fit preuve de son efficacité mais s'avéra trop astreignante et trop coûteuse pour être utilisée en routine. Des enzymes pancréatiques gastro-résistantes apparurent et il y eut des avancées majeures permettant un apport normal en graisse, et donc des apports énergétiques accrus chez la plupart des patients[44]. En 1978 un article supportant l'hypothèse qu'un bon état nutritionnel est associé à un meilleur pronostic parait[65]. C'est au cours de cette décennie qu'on décrit un traitement non chirurgical de l'iléus méconial par lavement à la Gastrografine[66], qu'on décrit le rôle pathogène de Burkholderia cepacia dont la dangerosité et la contagiosité entraîne des changements radicaux dans la pratique clinique et les habitudes sociales des patients[67] et qu'on se rend compte que sévérité de l'infection par P.aeruginosa et pronostic vital sont liés[68], observation à l'origine du protocole danois des antibiothérapies intraveineuses de 3 mois[69],[70],[71]. Les progrès sont si importants que la vision des médecins concernant la maladie change progressivement. En 1974 Crozier publie un article intitulé « La fibrose cystique - une maladie pas si fatale » [72]. En 1981 Norman annonce qu'une nouvelle ère commence et qu'il est « temps d'arrêter d'utiliser le terme de maladie génétique la plus létale » [73].
Dans les années 1980, de considérables avancées scientifiques ont lieu sur la compréhension de la maladie, mais sans grande repercussion sur la prise en charge des patients. En 1983 le diagnostic anténatal devient disponible pour les familles ayant déjà un enfant affecté. En 1984 un article de l'australien Peter Phelan met en évidence que les soins en centres spécialisés sont un des principaux facteurs de la meilleure survie observée chez les patients de Nouvelle-Galles du Sud par rapport à ceux d'Angleterre et du Pays de Galles, soignés dans des services de pédiatrie générale[74]. Cette observation fut suivie par la création de nombreux centres spécialisés dans la prise en charge de la mucoviscidose, notamment au Royaume-Uni. L'amélioration spectaculaire constatée après un traitement basique constitué de 2 semaines d'antibiothérapie intraveineuse, de physiothérapie, de support nutritionnel et de doses adéquates d'enzymes pancréatiques poussa de nombreux parents vers ces centres. Les antibiotiques intraveineux devinrent la pièce maitresse du traitement de l'infection chronique par P. aeruginosa qui touche virtuellement tous les patients[75],[76]. Devant la dégradation qui suit inévitablement l'arrêt de l'antibiothérapie, le centre spécialisé danois de Copenhague introduit des protocoles de cures trimestrielles systématiques de 2 semaines d'antibiothérapie intraveineuse, entraînant une amélioration impressionnante de la survie de leurs patients[70]. Afin d'améliorer la qualité de vie et de diminuer les couts, les traitements antibiotiques IV à domicile se developpent peu à peu. Après un article de 1981 de Margaret Hodson dans lequel elle décrit que des nébulisations biquotidienne de gentamicine et de carbenicilline permettent de stabiliser les patients ayant des infections à P. aeruginosa récidivantes[77], les nébulisations d'antibiotiques connaissent un regain d'intérêt important. En 1985 Littlewood note que des nébulisations de colomycine peuvent éradiquer une infection à P. aeruginosa[78] et empêcher sa chronicisation, jusqu'à la considérée comme inévitable. Une avancée majeure pour les patients au stade terminal de la maladie est la première transplantation cœur-poumon réalisée en 1984 à Harefield au Royaume-Uni et à Chapel Hill aux États-Unis[79]. Par la suite la translantation bi-pulmonaire devint plus populaire[80]. Des donneurs vivants ont été utilisés particulièrement aux États-Unis, en raison d'une pénurie de donneurs d'organes[81].
Cette décade connaît aussi d'importants progrès en matière de nutrition. Il devient évident que les patients avec un apport normal en graisse ont un meilleur apport énergétique, un meilleur état nutritionnel et une meilleures croissance[82]. Une autre avancée majeure est l'apparition de nouvelles préparations d'enzymes acido-resistantes, mélanges d'enzymes pancréatique de substitution ayant une activité amylolytique, lipolytique et proteolytique. La forme galénique d'enrobage gastro-résistant préserve l'activité enzymatique jusqu'à son arrivée dans le jéjunum. Ces préparations ont été commercialisées sous les noms de spécialité de Pancrease puis de Creon. Seul cette dernière spécialité est disponible en France (Solvay-Pharma propiétaire de la marque jusqu'en 2018).Il devient progressivement évident pour de nombreux praticiens que la restriction en graisse est maintenant rarement indiquée si des doses adéquates d'extraits pancréatiques sont apportées[83]. Dans les cas de dénutrition, si la nutrition intraveineuse parenterale s'avère être une solution sur le court terme, sa mise en œuvre n'est pas aisée et la nutrition entérale, par sonde naso-gastrique ou gastrostomie, prend progressivement de l'ampleur[44].Au début des années 1990, une vague d'enthousiasme suit l'identification du gène CFTR, certains patients pensant qu'un traitement curatif a été découvert. Des modèles animaux sont crées avec des souris mutantes permettant des recherches in vivo sur le fonctionnement du gène. Des études sur le remplacement génétique sont entreprises et même si les progrès sont lents, Davies envisage en 2001 que la thérapie génique sera une réalité dans les 5 à 10 ans[84]. Des traitements pharmacologiques alternatifs ou complémentaires destinés a améliorer la fonction CFTR sont également en cours d'étude[85]. Des centres spécialisés dans la prise en charge des adultes mucoviscidosiques apparaissent. Avec l'avènement de la médecine fondée sur les faits, tous les traitements, traditionnels ou récent, sont passés au crible. Antibiothérapie intraveineuse, bronchodilatateur et steroides inhalés semble utiles, le mucolytique Pulmozyme est efficace pour améliorer la fonction respiratoire, P. aeruginosa peut être éradiqué par un traitement précoce et l'infection chronique bénéficie d'une antibiothérapie inhalée au long court et de cures trimestrielles d'antibiotiques intraveineux. Les stéroïdes oraux s'avèrent trop toxique sur des durées prolongées[44]. Les différentes techniques de physiothérapie sont également réevaluées ce qui donne lieu en 2002 à une recommandation du CF Trust[86]. On met en évidence les risques de transmission d'agents infectieux tels que Burkholderia cepacia et Pseudomonas aeruginosa entre patients, ce qui entraîne une disparition des réunions et groupes de patients, modifiant profondement leur vie sociale[44].
Origine des mutations
Des auteurs ont tenté de déterminer l'âge et le lieu d'origine des principales mutations responsables de la mucoviscidose par analyse génétique de différentes populations. L'ancienneté de la mutation ΔF508, la plus fréquente, est controversée, avec des estimations comprises entre 3 000 ans[87] et plus de 50 000 ans[88]. En 2001 une étude a confirmé l'âge ancien de cette mutation, estimé entre 11 000 et 34 000 ans[89]; et une autre a estimé que l'apparition de ΔF508 pouvait être antérieure à l'expansion des hommes modernes[90]. Pré-néolithique, ΔF508 est donc une ancienne mutation dans l'histoire de l'Homme.
Les autres mutations retrouvées dans la mucoviscidose sont légèrement plus récentes[91].
La détermination, par analyse génétique de l'origine géographique des mutations responsables de la mucoviscidose, ne semble pas envisageable compte tenu du fait que ces mutations existaient probablement chez les ancêtres des Européens, avant même la genèse des populations actuelles; et que les modifications successives de ces populations ont probablement effacé les traces de l'origine géographique de ces mutations génétiques[92].
Épidémiologie
Dans la population caucasienne (ou europoïde), la mucoviscidose est la plus fréquente des maladies génétiques héréditaires graves[93]. Elle touche environ un enfant sur 2 500 naissances en Europe et en Amérique du Nord[3] et la fréquence des hétérozygotes, porteurs sains de la maladie, y est d’environ 1/25[2], soit 4% de la population générale occidentale.
La mucoviscidose est présente dans toutes les populations du globe, mais à des prévalences variables. Il existe ainsi très peu de cas chez les Noirs et encore moins chez les Asiatiques[94]. Respectivement l'incidence à la naissance - nombre de nouveau cas observée dans une période et une population donnée - et la fréquence des hétérozygotes est au Moyen-Orient d'environ 1/4 400 et de 1/33, dans la population Hispanique de 1/8 500 et 1/46, chez les Noirs américains et Africains de 1/20 000 et 1/70 et chez les Asiatiques de 1/32 400 et 1/90[95].
La prévalence - nombre de cas présents à un moment donné dans une population donnée - de la mucoviscidose, comme celle de toute maladie rare, est difficile à évaluer avec précision[96],[97]. La prévalence en Europe est estimée entre 8 et 12 cas pour 100 000 habitants[93],[96]. La Cystic Cibrosis Foundation estime le nombre de malade aux États-Unis d'Amérique à environ 30 000 enfants et adultes et à 70 000 le nombre total de malades dans le monde[5].
En France il existe depuis 2002 un dépistage néonatal systématique de la mucoviscidose ce qui a permit de réévaluer l’incidence de la maladie dans ce pays. Elle serait près de deux fois moins élevée que les estimations antérieures, d'environ un nouveau-né sur 4 600 naissances, soit une fréquence d'hétérozygotes de l’ordre de 1 sur 35[98]. L'incidence varie selon les régions de 1/2 500 dans le nord-ouest à 1/10 000 dans le sud-est[2]. Il y aurait environ 5 à 6 000 personnes atteintes de la mucoviscidose[4] et 2 millions de porteurs hétérozygotes sains.
Le sex-ratio est proche de 1 ce qui signifie que les hommes et les femmes sont atteints avec la même fréquence par la maladie. Par contre l'atteinte est souvent plus grave chez les femmes[99],[100],[101].
- Hypothèses concernant la prévalence élevée de la mucoviscidose
La fréquence élevée d'hétérozygotes porteurs sains, d’environ 1/25[2], soit 4% de la population occidentale, peut surprendre et pourrait être expliquée par le fait que l'état hétérozygote augmenterait la résistance contre certaines maladies. Cet avantage sélectif au niveau de l'état hétérozygote a été retrouvé par exemple chez les drépanocytaires chez qui le portage du gène Hbs atténue l'expression et la gravité de l'accès palustre[102]. Des résistances à différentes maladies infectieuses ont été proposées comme étant la cause de l'avantage hétérozygote lié au gène CF :
- La toxine du choléra ayant besoin de la protéine CFTR pour fonctionner correctement, des auteurs ont proposé l'hypothèse que les porteurs hétérozygotes du gène CF bénéficient d'une résistance accrue vis à vis du choléra ou d'autres formes de diarrhée infectieuse[103]. Mais cette hypothèse n'a pas été confirmée par la suite[104],[105].
- L'entrée de Salmonella enterica typhi, agent responsable de la fièvre typhoïde, dans les cellule nécessite la présence de protéines CFTR normales[106]., ce qui pourrait faire penser que les porteurs d'un gène mutant puissent être protégés contre cette maladie. Aucune étude in vivo n'a encore confirmé cette hypothèse.
- D'autres auteurs, sur la base de modèles génétiques, démographiques et épidémiologiques, ont fait remarquer que les épidémies de choléra ou de fièvre typhoïde n'avaient pas pu exercer assez de pression sélective pour donner les chiffres actuels de prévalence de la mutation. Et font remarquer que la tuberculose a pu exercer une telle pression[107].
Étiologie et physiopathologie
Le gène responsable de la mucoviscidose
Le gène CFTR, Cystic fibrosis transmembrane conductance regulator, code la synthèse d'une protéine appelée CFTR dont la fonction est essentielle à l'organisme. Les mutations dans ce gène sont responsables de la mucoviscidose et de l'aplasie congénitale du canal déférent. C'est un grand gène constitué d'environ 250 000 paires de bases répartis en 27 exons et codant un ARNm de 6,5 kb. Il est localisé sur le locus 7q31.2, dans la région q31.2 du bras long du chromosome 7 [108],[109],[110].
Mutations du gène CFTR
On dénombre en 2007 plus de 1 500 mutations du gène CFTR[1].
La majorité des mutations du gène CFTR sont des mutations ponctuelles. Les plus fréquentes sont des mutations faux-sens (42%), puis viennent des microinsertions et des microdélétions (24%), des mutations non-sens (16%), des mutations d'épissage (16%) et enfin des délétions d'un acide aminé (2%). On rapporte également quelques grandes délétions[111].
Les mutations du gène CFTR sont regroupées en 6 classes en fonction des conséquences fonctionnelles qu'elles occasionnent. Certaines mutations entraînent des anomalies quantitatives ou qualitatives sur la protéine CFTR [111]:
- Classe 1 : mutations altérant la production de la protéine CFTR.
- Classe 2 : mutations perturbant le processus de maturation cellulaire de la protéine CFTR.
- Classe 3 : mutations perturbant la régulation du canal chlorure.
- Classe 4 : mutations altérant la conduction du canal chlorure.
- Classe 5 : mutations altérant la stabilité de l'ARNm CFTR.
- Classe 6 : mutations altérant la stabilité de la protéine mature.
La mutation la plus fréquente est la mutation Delta F508 (ΔF508) qui consiste en une délétion de trois nucléotides au niveau du dixième exon du gène, aboutissant à l'élimination d'un acide aminé, la phénylalanine, en position 508. Cette mutation est retrouvée dans près de deux tiers des cas avec des variations importantes selon les populations étudiées, ainsi on retrouve un gradient de répartition suivant une direction nord-ouest/sud-est en Europe, avec par exemple 88% de DF508 au Danemark, 81% en Bretagne et 50% en Italie[111]. Seule quatre autres mutations, hors ΔF508, représentent plus de 1% des cas, il s'agit de G542X, G551D, N1303K et W1282X[1]. Toutes les autres mutations sont rares, voire exceptionnelles, uniquement retrouvées au sein d'une seule famille.
Les mutations les plus fréquentes dans les populations caucasiennes sont [1] :
Nom de la mutation Fréquence Nom de la mutation Fréquence ΔF508 66.0 % R553X 0.7 % G542X 2.4 % 621+1G 0.7 % G551D 1.6 % 1717-1G 0.6 % N1303K 1.3 % R117H 0.3 % W1282X 1.2 % R1162X 0.33 % Les génotypes retrouvés sont [111]:
Paire de mutations Fréquence ΔF508 - ΔF508 (homozygote) 50% ΔF508 - autre mutation (hétérozygote composite) 40% autre mutation - autre mutation 10% Corrélation génotype et phénotype
Il existe un certain rapport entre la mutation (le génotype) et les manifestations cliniques de la maladie (le phénotype). La très grande variabilité des manifestations et de la gravité de cette maladie est en rapport avec les multiples mutations du gène CFTR.
La pénétrance est habituellement de 100% chez les homozygotes avec mutations sévères mais la sévérité de la maladie reste variable avec des formes atténuées sans insuffisance pancréatique ou avec atteinte respiratoire modérée.
L'état homozygote ΔF508/ΔF508 est associée à la forme classique de la maladie avec une augmentation des électrolytes dans la sueur, une insuffisance pancréatique et une atteinte pulmonaire souvent sévère. Certaines mutations entraînent plus de troubles fonctionnelles que d'autres ainsi les études épidémiologiques montrent une mortalité différente dans des populations ayant des mutations différentes. Ainsi les mutations ΔF508/R117H, ΔF508/DeltaI507, ΔF508/3849+10kbC-→T et ΔF508/2789+5G-→A ont un taux de mortalité plus bas que les homozygotes DeltaF508. Et les mutations ΔF508/R117H, ΔF508/DeltaI507, ΔF508/ 3849+10 kbC-→T, ΔF508/2789+5G-→A et ΔF508/A455E ont des manifestations phénotypiques atténuées[112].
C'est pour la fonction pancréatique que la corrélation génotype/phénotype est la plus évidente, le pancréas peut présenter des lésions de gravité très variable selon les mutations, alors que la sévérité de l'atteinte pulmonaire peut être très différente chez des sujets ayant un même génotype CFTR, même au sein d'une même famille. Cette variabilité phénotypique de l'atteinte respiratoire pourrait être due à des facteurs génétiques distincts du gène CFTR ou à des facteurs environnementaux[113]. Ainsi les hétérozygotes ΔF508/A455E ont une meilleure fonction pulmonaire que les homozygotes ΔF508; cette mutation étant associée à une fonction pancréatique conservée, la moindre sévérité de l'atteinte pulmonaire pourrait être la conséquence d'un meilleur état nutritionnel[114].
La protéine CFTR
La protéine CFTR (cystic fibrosis transmembrane conductance regulator) est codée par le gène CFTR. C'est une protéine membranaire de 1 480 acides aminés et son poids moléculaire est de 168 kD[108],[109].
La molécule CFTR est composée de 5 domaines, deux domaines hydrophobe transmembranaires (membrane-spanning domains, MSD) comportant chacun six segments transmembranaire en hélice alpha, deux domaines hydrophile d'interaction avec les nucléotides (nucleotide-binding domains, NBD), et un domaine cytoplasmique de régulation (regulatory domain, R) codé par l'exon 13 et contenant de nombreux résidus chargés et la majorité des sites potentiels de phosphorylation [115].
La molécule se situe au pôle apical des cellules épithéliales et se comporte comme un canal ionique laissant passer l'ion chlorure selon un gradient électrochimique (transport actif secondaire, tirant son énergie du gradient électrochimique favorable à l'entrée de l'ion sodium dans la cellule entretenu par l'activité de la pompe sodium/potassium). L'activation du canal dépend de multiples phénomènes de phosphorylation du domaine de régulation (R) et de l'hydrolyse d'une molécule d'Adénosine triphosphate (ATP) sur les domaines d'interaction avec les nucléotides. L'hydrolyse du NBD en situation N-terminal permet l'ouverture du canal transmembranaire sélectif aux ions chlorure. L'hydrolyse de l'ATP sur le domaine NBD C-terminal, conduit à sa fermeture[116].
Outre sa fonction canal chlore, la protéine CFTR régule également d'autres canaux comme le canal chlore à rectification sortante, le canal sodium épithélial, et des canaux potassium à rectification entrante. D'autres fonctions sans rapport avec la régulation des canaux ont aussi été décrites comme le transport d'ATP, la modulation des phénomènes d'exocytose et endocytose, la régulation du pH des organelles intracellulaires. Ce qui fait de la protéine CFTR une protéine multi-fonctionnelle[111].
 La protéine membranaire CFTR avec marquage du site de la mutation Delta F508. Constituée de 5 domaines, elle se comporte comme un canal ionique laissant passer l'ion chlorure mais possède également d'autres fonctions.
La protéine membranaire CFTR avec marquage du site de la mutation Delta F508. Constituée de 5 domaines, elle se comporte comme un canal ionique laissant passer l'ion chlorure mais possède également d'autres fonctions.Physiopathologie des manifestations
La mutation du gène de la mucoviscidose entraîne un défaut dans la synthèse de la protéine cystic fibrosis transmenbrane conductance regulator ou CFTR (famille ATP Binding Cassette), qui est une protéine membranaire entrant dans la formation d'un canal ionique sélectif aux anions et principalement chlorures.
Le défaut de synthèse de la protéine entraîne un déficit en chlore extracellulaire et donc un défaut d'hydratation du mucus, une hyperviscosité des sécrétions épithéliales liée à une réabsorption exagérée de l'eau secondaire à la rétention cellulaire de l'ion chlore.
L'insuffisance de fonctionnement des glandes exocrines se remarque surtout au niveau du poumon, du pancréas et du foie. Les atteintes digestives sont les plus précoces et les premières historiquement décrites.
Au niveau du pancréas
Les enzymes du pancréas, comme la lipase et la trypsine, qui servent à la digestion sont mal excrétées dans la lumière intestinale parce que les canaux pancréatiques se bouchent du fait d'une sécrétion trop épaisse et d'une fibrose importante. Les enzymes agressent alors directement le tissu pancréatique, contribuant à la fibrose. Des kystes peuvent éventuellement se former. Lorsque les fonctions pancréatiques sont significativement altérées, on parle d'insuffisance pancréatique[117].
Le pancréas est atteint dans sa fonction exocrine et endocrine de deux manières :
- atteinte de la fonction exocrine : la réduction ou l'absence d'arrivée d'enzymes de digestion dans la lumière intestinale conduit à un syndrome de malabsorption pouvant avoir un retentissement sur le développement staturo-pondéral et pubertaire des enfants.
- atteinte de la fonction endocrine : la destruction des cellules bêta des îlots de Langerhans conduit à un défaut de sécrétion d'insuline et un diabète insulinoprive - mais bien distinct du diabète de type 1 auto-immun - apparait. Si l'insuffisance pancréatique exocrine apparaît précocement, les îlots de Langerhans sont longtemps épargnés. Il semble que la destruction des îlots ne soit pas causée uniquement par le développement progressif d'une fibrose pancréatique mais aussi par d'autres phénomènes tels qu'une ischémie. La capacité à sécréter de l’insuline reste longtemps normale et plusieurs plusieurs années s'écoulent entre la période d'intolérance au glucose et l'apparition du diabète. Outre les anomalies de sécrétion de l'insuline, d'autres facteurs pourraient contribuer développement du diabète tels qu'une insulino-résistance transitoire[118].
Au niveau hépatobiliaire
La viscosité de la bile est augmentée par les dysfonctionnements de la protéine CFTR des cellules épithéliales biliaires. Les canaux qui transportent la bile vont se boucher à cause de ce liquide trop épais. La répétition de ces obstructions conduit à des phénomènes de cirrhose localisée et d'hépatomégalie.
Les conséquences habituelles de la cirrhose biliaire sont les mêmes chez le patient mucoviscidosique que dans le reste de la population. L'hypertension dans la veine porte, les hémorragies digestives et l'insuffisance hépatocellulaire peuvent conduire à la greffe hépatique.
Au niveau du poumon
Le liquide de surface tapissant l'arbre bronchique se compose d'eau et de mucus. Les canaux CFTR servent à la sécrétion active de chlore vers ce liquide, ils interagissent avec les canaux d'absorption du sodium pour limiter leur travail. Ce mouvement de chlore entraîne un mouvement de sodium et d'eau. Cette sécrétion d'eau permet d'hydrater le liquide de surface bronchique et de lui maintenir des propriétés rhéologiques adéquates pour une clairance muco-ciliaire efficace. La clairance muco-ciliaire est le débit de liquide de surface transporté par les cellules ciliées de l'arbre bronchique. L'évacuation de ce liquide permet l'élimination des poussières et des agents infectieux vers le système digestif.
L'altération fonctionnelle des canaux CFTR entraîne une déshydratation du liquide de surface bronchique. Les modifications des propriétés du mucus, et notamment l'augmentation de sa viscosité rendant plus difficile son évacuation par les cils, conduit à une obstruction chronique des bronches ainsi qu'à la non évacuation des poussières et bactéries. De plus les propriétés antibactériennes du mucus sont diminuées. Tous ces éléments favorisent l'apparition d'une infection précoce devenant rapidement chronique associée à une réaction inflammatoire marquée[119].
Au fil du temps, différentes bactéries colonisent les voies respiratoire d'un même malade. Une large étude[120] publiée en 2000 faite sur 1 000 enfants de moins de 2 ans retrouvait sur des prélèvements bronchoscopiques 19 % d'Haemophilus influenzae, 42 % de Staphylococcus aureus, 29 % de Pseudomonas aeruginosa, 7 % de Stenotrophomonas maltophilia et moins de 1 % de Burkholderia cepacia. D’autres agents infectieux, bactériens, viraux ou fongiques, peuvent parfois être retrouvés. Initialement ce sont des bactéries banales telles que Haemophilus influenzae et Staphylococcus aureus qui colonisent et infectent les poumons, puis survient la colonisation à Pseudomonas aeruginosa, qui constitue un tournant dans l’évolution de la maladie respiratoire[119]. L'utilisation répétée d'antibiotiques mais aussi d'autres facteurs encore indéterminés et liées à la maladie elle-même, favorisent la colonisation du tractus bronchique par des bacilles pyocyaniques généralement résistants aux antibiotiques d'utilisation courante. Ces bactéries sont exceptionnelles chez les personnes indemnes de mucoviscidose[120].
Il existe une inflammation de l'épithélium bronchique dont les mécanismes sont incomplètement élucidés. Il n'est pas encore tranché que cette inflammation soit directement consécutive à la colonisation bactérienne du mucus. Les études physiopathologiques se concentrent sur les autres fonctions de la protéine CFTR, qui aurait un rôle dans la régulation de l'inflammation, voire un rôle dans la destruction de Pseudomonas Aeruginosa.
À terme, l'inflammation et l'infection chronique entretiennent un cercle vicieux et entraînent une dégradation pulmonaire par des lésions du tissu pulmonaire conduisant à un tableau de broncho-pneumopathie chronique obstructive.
Description clinique
La mucoviscidose est une maladie du tissu épithélial des glandes exocrines de l'organisme. Ce tissu se retrouve dans de nombreux organes comme les voies respiratoires (bronches, cavités nasales…), le pancréas, le foie et l'arbre biliaire, l'intestin grêle, les canaux déférents des organes génitaux masculins, les glandes sudoripares de la peau.
La sévérité du tableau clinique varie selon les mutations du gène CFTR, mais aussi selon d'autres facteurs génétiques et environnementaux. L'histoire naturelle de la maladie est donc très variable et va du décès précoce dans la petite enfance par aggravation progressive de l'obstruction pulmonaire avec bronchectasie, à une insuffisance pancréatique avec atteinte obstructive pulmonaire progressive durant l'adolescence et une augmentation de la fréquence des hospitalisations suite à l'atteinte respiratoire au début de l'âge adulte[114].
La forme clinique la plus fréquente associe troubles respiratoires (bronchite chronique), troubles digestifs (stéatorrhée, constipation) et troubles de la croissance staturopondérale[93].
Manifestations pulmonaires
Les manifestations pulmonaires restent la cause majeure de la morbidité et de la mortalité. Il existe une inflammation chronique des bronches avec surinfection bactérienne qui entretiennent un cercle vicieux et sont la cause de la dégradation de l'état pulmonaire. Les premiers germes colonisant les voies aériennes sont Haemophilus influenzae et Staphylococcus aureus, puis Pseudomonas aeruginosa (bacille pyocyanique) à un stade plus avancé[114],[119].
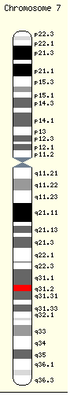
 Hippocratisme digital majeur
Hippocratisme digital majeurL'histoire naturelle de la maladie pulmonaire est une dégradation progressive de l'état respiratoire, évoluant par poussées, débutant par des symptômes tels qu'une toux et des crachats et aboutissant à une insuffisance respiratoire grave voir à une cœur pulmonaire chronique[114],[119],[121],[122].
Les premières manifestations respiratoires sont une toux chronique ou récurrente — initialement sèche — parfois paroxystique et pouvant entraîner des vomissements, puis grasse et éventuellement purulente, des crachats et des difficultés expiratoires qui surviennent lorsque la fonction respiratoire se détériore. Des épisodes de bronchites chroniques surviennent chez l'enfant et entraîne progressivement des lésions de l'arbre bronchique menant à des bronchectasies.
Une fois l'atteinte respiratoire bien installée, les malades peuvent présenter des douleurs thoraciques, des pneumopathies récurrentes, des formes atypiques d'asthme et un wheezing récurrent. Des épisodes de pneumothorax ou d'hémoptysies peuvent engager le pronostic vital. Toutes ces manifestations sont des complications pulmonaires et peuvent parfois être les manifestations initiales révélatrices de la maladie.
Les lésions des poumons s'aggravent progressivement, avec formation d'abcès et de kystes et fibrose du parenchyme pulmonaire proches des voies aériennes, aboutissant à une insuffisance respiratoire majeure.
À l'examen clinique du patient, on peut retrouver un hippocratisme digital ou une dystrophie thoracique d'apparition précoce et témoignant de la bronchopathie chronique obstructive[122]. L'auscultation est variable selon l'atteinte pulmonaire, elle peut être normale ou retrouver des sibilants ou des râles bonchiques.
À la radiographie pulmonaire on retrouve des images bronchiques et alvéolaires non spécifiques comme un épaississement péribronchique, une distension pulmonaire avec emphysème, une atélectasie, un foyer alvéolaire, des signes de bronchectasies[122].
Manifestations digestives
Manifestations intestinales
Les manifestations surviennent dès la période néo-natale sous forme d'un iléus méconial qui survient chez 10 à 20 % des enfants porteurs de la maladie et peut être diagnostiqué lors de l'échographie morphologique montrant l'existence d'une hyperéchogénicité intestinale[123]. Cet iléus méconial correspond à une occlusion intestinale aiguë néonatale basse qui se traduit chez le nouveau né par l'absence d'émission de méconium associé à des vomissements et à des ballonnements. Le lavement permet l'évacuation d'un méconium obstructif visqueux[122].
Le syndrome de l'occlusion intestinale distale (SOID), qui touche plus volontiers l'enfant, est une occlusion de l'intestin grêle qui mime la crise appendiculaire.
Le reflux gastro-œsophagien est fréquent chez le nourrisson, parfois majoré par les manœuvres de kinésithérapie respiratoire et la toux chronique. Ce reflux ne s'amende pas avec l'âge de l'enfant à l'inverse de la majorité des reflux gastro-œsophagiens.
Le prolapsus rectal est également plus fréquent chez les nourrissons mucoviscidosiques et justifie la réalisation d'un test de la sueur. Il est favorisé par un volume de selles plus important, la malnutrition et l'augmentation de pression intra abdominale lors des efforts de toux et de défécation[124].
Une sténose colique peut se voir lors d'un surdosage du traitement par enzymes pancréatiques. Une hémorragie digestive complique les manifestations hépato-biliaires.
Manifestations hépato biliaires
 Éléments anatomiques de l'arbre biliaire
Éléments anatomiques de l'arbre biliaireL'atteinte hépato-biliaire de la mucoviscidose est fréquente mais n'évolue vers la cirrhose biliaire que dans 10 à 15 % des cas[122]. Elle se traduit par une élévation de la viscosité de la bile puis par une obstruction à l'évacuation de cette dernière dans les canaux hépatiques vers la vésicule biliaire. La conséquence est l'apparition d'un ictère rétentionnel néonatal (coloration jaune de la peau et des muqueuses) qui est le reflet de l'élévation de la bilirubine non conjuguée dans le sang. Biologiquement on note l'élévation concomitante des transaminases et des γGT.
La cirrhose biliaire multifocale se complique de la même manière qu'une cirrhose commune avec l'apparition d'une hypertension portale et d'hémorragies digestives, d'une insuffisance hépato-cellulaire et d'épisodes de décompensation oedémato-ascitique.
La vésicule biliaire est souvent atrophique et on peut parfois retrouver également une lithiase vésiculaire, le plus souvent asymptomatique mais pouvant aggraver une pathologie pancréatique sous-jacente.
Manifestations pancréatiques
L'insuffisance pancréatique exocrine est présente chez la grande majorité des patients, dans environ 90 % des cas[122]. Elle est responsable d'un défaut d'absorption des graisses (acides gras essentiels), des protéines et des vitamines liposolubles (A, D, E, K, et parfois hydrosoluble comme B12). La malabsorption des graisse entraîne une diarrhée chronique graisseuse (caractérisant la stéatorrhée) avec ballonnement abdominal et appétit conservé qui contraste avec l'hypotrophie pondérale puis staturale. Le trouble de l'absorption de la vitamine D entraîne une mauvaise minéralisation des os et un risque de rachitisme et de fracture. Parmi les autres manifestations de la malabsorption, on retrouve une insuffisance de croissance, un retard pubertaire, une anémie, des troubles carentiels (en vitamines, oligo-éléments et minéraux).
On peut voir parfois l'apparition de pancréatite aigüe ou chronique, plus fréquemment chez les patients avec une fonction pancréatique conservée[114].
Un diabète insulinoprive, traduit une atteinte de la fonction endocrine du pancréas, secondaire à la destruction des îlots de Langerhans par la fibrose et à des anomalies fonctionnelles de la sécrétion d’insuline. Ce diabète est différent de du diabète de type 1 auto-immun. Il touche entre 13 et 16 % des malades, sa prévalence augmentant avec l’âge et le sexe féminin. L'âge médian de découverte est de 20 ans. Il ne se développe pas chez les patients sans insuffisance pancréatique[118].
Manifestations génitales
Les manifestations génitales de la mucoviscidose posent un problème de plus en plus important avec l'amélioration de la survie et le désir d'enfant des patients[122].
Chez la femme on retrouve une hypofertilité due à des modifications de la glaire cervicale, néanmoins les grossesses de femmes mucoviscidosiques ne sont plus rares.
Chez l'homme on retrouve une stérilité par atrésie bilatérale des canaux déférents. La découverte de cette anomalie lors de l'investigation d'une infertilité d'un couple doit d'ailleurs faire rechercher systématiquement cette pathologie. L'atrophie ou hypoplasie des voies excrétrices des spermatozoïdes (canal déférent et vésicules séminales) entraîne une azoospermie ou oligospermie sévère (moins de 5 millions de spermatozoïdes par ml). La composition du sperme est aussi modifiée[125] :
Normal Mucoviscidose pH >8 <7 Acide citrique 400-1500 mg/100 ml > 2000 mg/100 ml Phosphatase acide 140-290 microg/ml 760-1140 microg/ml Fructose 250-720 mg/100ml 30–80 mg/100ml Autres manifestations
Les affections rhino-sinusiennes sont fréquentes. La qualité du mucus dans les sinus est la même que celle des bronches. Une inflammation et une infection chronique entraîne une sinusite chronique et secondairement une polypose nasale[126],[127].
Les désordres hydro-électrolytiques avec déshydratation par perte de sel peuvent apparaitre lors d'efforts ou de forte chaleur. Le diagnostic de mucoviscidose devant un tableau de déshydratation avec hyponatrémie, hypochlorémie et alcalose métabolique n'est pas rare notamment l'été[128].
Une myocardiopathie probablement d'origine carentielle peut être décrite[129] ainsi qu'une hypertension artérielle pulmonaire consécutive à un cœur pulmonaire chronique.
Diagnostic et dépistage
Diagnostic
La suspicion de mucoviscidose se base sur les signes cliniques suivants mais cette liste n'est pas limitée :
- Des maladies pulmonaires ou bronchiques chroniques, récidivantes (toux chronique, encombrement et syndrome obstructif chronique, colonisation par certains germes, anomalies radiologiques, sinusites chroniques, hippocratisme digital)
- Des troubles digestifs (iléus méconial, prolapsus rectal, insuffisance pancréatique et syndrome de malabsorption, maladies hépatobiliaires chroniques)
- Une azoospermie,
- Un syndrome de perte de sel…
Le diagnostic est établi chez une personne présentant un ou plus des signes cliniques précédents associés à au moins un des signes de mauvais fonctionnement du gène CFTR suivants :
- Présence de mutation anormale du gène CFTR sur chacun des chromosomes 7, ou
- Deux tests de la sueur positifs (>60 mEq/L), ou
- Une recherche de différence de potentiel de l'épithélium nasal positive.
En l'absence de signe clinique, le diagnostic peut être fait :
- si un enfant est porteur de la même mutation du gène CFTR sur chacun des chromosomes 7 qu'un de ses frères et sœurs malades de la mucoviscidose,
- au cours d'un dépistage néo natal systématique (en 2002, 12,8 % des mucoviscidoses sont diagnostiquées de cette façon aux États-Unis),
- au cours d'un dépistage in utero (en 2002, 4 % des mucoviscidoses sont diagnostiquées pendant la période prénatale aux États-Unis).
Test de la sueur
Le test de la sueur est le dosage du chlore contenu dans la sueur du patient et représente un des principaux test diagnostique de la mucoviscidose. Il comprend trois temps : l'obtention de la sueur, le recueil de la sueur et la détermination de la concentration en chlorure[131].
La méthode de référence pour obtenir de la sueur est la stimulation des glandes sudoripares, au niveau de la face interne du bras, au-dessus du pli du coude, par iontophorèse à la pilocarpine[132]: le passage d’un faible courant à travers la peau entre deux électrodes permet de faire passer la pilocarpine - molécule possédant des propriétés cholinergiques - à travers la peau, stimulant ainsi la sécrétion de sueur par les glandes sudoripares.
La méthode de référence pour le recueil de la sueur, délicat, est le recueil sur papier filtre[132], décrite par Gibson et Cooke[30]. Il faut recueillir un échantillon d'au moins 100 mg de sueur. L'âge minimum requis est de 5 semaines avec un poids corporel d'environ 4 kg[132]. Cet examen indolore dure 15 à 30 minutes.
Les méthodes utilisées pour déterminer la concentration en chlorures de la sueur sont de trois catégories : les méthodes quantitatives qui mesurent avec précision le poids de la sueur recueillie, les méthodes semi-quantitatives qui déterminent la concentration en électrolytes avec une approximation du volume recueilli et les méthodes qualitatives basées sur des réactions chimiques de précipitation permettent une estimation qualitative de la concentration en chlorures[132].
Chez les malades, les glandes sudoripares ont une teneur augmentée en ions Na+, K+ et Cl-. On ne teste cependant que la teneur en chlorure, plus discriminative que la teneur en sodium pour poser le diagnostic[133].
Un test de la sueur retrouvant une concentration de chlore supérieure a 60 mmol/l (millimoles par litre) est dit positif et est en faveur du diagnostic de mucoviscidose, il doit être positif à deux reprises pour affirmer le diagnostic. Un test retrouvant une concentration de chlore intermédiaire comprise entre 40 mmol/l et 60 mmol/l est douteux et suggère le diagnostic sans pour autant le poser avec certitude, des examens complémentaires sont nécessaires. Une concentration inférieure à 40 mmol/l est normale et le risque de mucoviscidose est faible.
Ce test est sensible, positif plus de 90% des cas de mucoviscidose[114]. Il n'y a pas de corrélation entre la valeur retrouvée lors du test et la gravité de l'atteinte[134]. Le test ne permet pas d'identifier les sujets hétérozygotes qui transmettent la maladie même si ceux-ci ont statistiquement des concentrations intermédiaires.
Les résultats supérieurs à 160 mmol/l ne sont pas physiologiquement possibles et sont à rapporter avec un problème technique[114].
Les faux positifs - le test est positif mais la personne n'est pas malade - exceptionnels, peuvent se retrouver dans le syndrome de Hurler, la fucosidose, la glycogénose de type 1, l'insuffisance surrénalienne aiguë, un déficit en alpha 1-antitrypsine, une hypothyroïdie, une dysplasie ectodermique, un diabète insipide, une néphronophtise[135], une anorexie, une dysfonction du système nerveux autonome, une maladie coeliaque, une cholestase familiale, une hypogammaglobulinémie, une hypoparathyroïdie, un syndrome de Klinefelter, une malnutrition, une mucopolysaccharidose de type 1, un pseudohypoaldosternonisme[130].
À l'inverse il existe de rares cas de mucoviscidose avec un test de la sueur douteux ou négatif. Quelques mutations sont associées à des résultats limites telles que R117H, 3849 + 10kb C-T, R334W ou P67L[136],[137],[138]. Très rarement, le test peut même être normal chez un patient présentant un génotype mucoviscidosique avéré[139],[140],[141],[142]. De plus environ 1 à 2 % des malades ont une concentration de chlore inférieure à 60 mmol/l[143]. Dans cette situation, seule l'identification de deux mutations sur le gène CFTR permet un diagnostic de certitude.
Différence de potentiel de l'épithélium nasal
Cet examen proposé par Knowles[144] puis standardisé[145],[146] consiste a mesurer la différence de potentiel (DDP) de l'épithélium nasal et permet l'exploration in vivo des transports ioniques transépithéliaux. Il peut être utilisé à partir de 6 ans et il est réalisé dans de nombreux centres dans le monde dans le cadre de la démarche diagnostique. La mesure de la DDP nasal est accompagnée par des tests pharmacologiques associés destinées à mieux différencier la population malade de la non malade.
La DDP nasal n'est pas corrélée avec le test de la sueur ou avec la sévérité de la maladie ; et cette méthode ne détecte pas les sujets hétérozygotes[147]. La valeur basale de la DDP nasal est modifiée par des pathologies telles l'inflammation, l'infection ou la polypose nasale[147],[148].
Les individus atteints de mucoviscidose comparés à des individus indemnes ont[114] :
- Une différence de potentiel élevée (plus négative) reflétant une absorption accrue du sodium,
- Une variation plus importante de la différence de potentiel durant l'administration au niveau de la muqueuse nasale d'amiloride, un inhibiteur de l'absorption sodique,
- Des variations minimes de la différence de potentiel sous perfusion de d'amiloride, de solution sans chlore et d'un agoniste ß2.
La mesure de la DDP nasal avec stimulations pharmacologiques dans le bilan de la mucoviscidose n'a pas d'intérêt dans les formes typiques de la maladie mais elle est intéressante dans les formes cliniques atypiques associées à des tests de la sueur « limites »[149].
Génotypage
Le génotypage par analyse génétique à partir d'un échantillon de sang vise à rechercher la présence d'une mutation anormale sur le gène CFTR. Il est essentiellement utilisé lors de la démarche diagnostique lorsque le test à la sueur n'est pas réalisable ou non informatif. Il peut être utilisé en première intention dans les cas suivants [114]:
- Dépistage prénatal chez un fœtus à haut risque,
- Dépistage prénatal chez un fœtus à faible risque mais avec une hyperéchogénicité digestive fœtale,
- Dépistage néonatal,
- Enfants symptomatiques (avec un iléus méconial) trop jeunes (moins de 5 semaines) pour pouvoir réaliser un test de la sueur (impossibilité de collecter une quantité suffisante de sueur),
- Dépistage de la fratrie d'un patient atteint chez qui on a identifié les deux mutations du gène CFTR.
L’analyse génétique par la méthode de détection ciblée recherche les mutations parmi les plus fréquentes. En 2004, l'American College of Medical Genetics (ACMG) recommandait des kits analysant la présence de 23 mutations parmi les plus fréquentes, permettant d'atteindre un taux de détection allant de 57% à 97% selon les populations étudiées[114]. D'autres méthodes telles la recherche de réarrangements, délétions, insertions, duplications ou la méthodes de balayage d’exons permettent de compléter l'analyse.
Chez un homme avec azoospermie ou oligospermie
La recherche d'une hypoplasie voire une agénésie des vésicules séminales et/ou des déférents par un urologue ou par d'imagerie médicale est la première étape. Si celle-ci est positive la recherche de mutation du gène CFTR est réalisée.
Diagnostic différentiel
Face à des éléments cliniques évoquant la mucoviscidose, se pose le problème d'un diagnostic différentiel avec d'autres maladies telles que :
- Une dilatation des bronches due à d'autres étiologies telles qu'un déficit immunitaire, un syndrome d'immobilité ciliaire, des séquelles de virose pulmonaire sévère (adénovirus), une pneumopathie négligée (tuberculose), une dysplasie broncho-pulmonaire ;
- Un wheezing chronique, un asthme[150],[151] ;
- Un tabagisme passif pouvant entraîner des manifestations respiratoires peu spécifiques ;
- Une maladie de Hirschsprung ou un syndrome du bouchon méconial, dans les formes de mucoviscidose avec occlusion intestinale néonatale associée[152],[153] ;
- Un déficit en alpha-1-antitrypsine, avec emphysème pulmonaire et signes respiratoires ;
- Un syndrome de dyskinésie ciliaire dont la maladie de Kartagener, exceptionnelle ;
- Un syndrome de Shwachman-Diamond[154] ;
- Des pancréatites aiguës récidivantes[155],[156] ;
- Une insuffisance pancréatique chronique[157].
Dépistage
Dépistage néo-natal systématique
Les arguments en faveur d'un dépistage systématique sont:
- L'existence d'un test fiable,
- Le diagnostic parfois tardif de la maladie en raison des formes frustres,
- Le bénéfice d'un diagnostic et d'une prise en charge précoce des enfants atteints : plus la prise en charge - nutritionnelle essentiellement - est précoce, meilleur est le pronostic.
Il se déroule en deux étapes [158],[159] :
- Le test consiste à doser la trypsine immuno–réactive (TIR), proenzyme sécrétée par le pancréas circulant dans le sang. Il est réalisé trois jours après la naissance de l'enfant à partir d'une goutte de sang séchée en même temps que le dépistage de la phénylcétonurie, de l'hypothyroïdie et de l'hyperplasie congénitale des surrénales.
- La détection d'un niveau anormalement élevé de trypsine (>60 μg/l) conduit à réaliser un diagnostic par génotypage à la recherche des mutations CFTR les plus fréquentes. Le test génétique nécessite au préalable l'accord écrit et signé des parents (en France la loi traitant de l’analyse de l’ADN à des fins médicales est le Décret n° 2000-570 du 23 Juin 2000[160]).
Les enfants chez lesquels une anomalie génétique (une ou deux mutation CFTR) est mise en évidence subiront dans le mois suivant leur naissance un test à la sueur pour déterminer leur statut de malade ou de porteur hétérozygote. Si aucune mutation n’est retrouvée, un contrôle de la trypsine est fait à J21.
Tous les programmes de dépistage utilisent maintenant ce test couplé TIR-ADN. La recherche des mutations comprend au minimum la mutation ΔF508 mais utilise le plus souvent des kits avec 20, 30 voire 50 mutations, représentant plus de 90 % des mutations existantes[159]. Ces programmes de dépistage ne sont pas encore figés et sont évolutifs afin de devenir le plus efficace possible.
- En France
La France a été le premier pays à instaurer un programme de dépistage néonatal systématique fin 2002, dans les 3 premiers jours de vie à la maternité[158]. Entre fin 2002 et fin 2005, 2 717 992 nouveau-nés ont bénéficié de ce programme. Une TIR élevée a été retrouvée chez 18 610 nouveau-nés (soit 0,7 % du total) ; 18 054 (soit 98 % d’entre-eux) ont bénéficié d'une recherche des principales mutations du gène CFTR ; 3 469 enfants (soit 1,3 pour 1000 nouveau-nés testés) ont été adressés à un CRCM (Centre de Références et de Compétences de la Mucoviscidose) parce qu’ils avaient 1 ou 2 mutations (n=2003) ou parce qu’ils avaient une hypertrypsinémie persistante à J21 (n=1466) ; 621 mucoviscidoses ont ainsi été diagnostiquées soit une incidence de 1 pour 4376 naissances, très variables selon les régions (de 1/2749 en Bretagne pour la plus forte à 1/7077 en Midi-Pyrénées pour la plus faible). Le kit CF30 détectait 87 % des mutations[159].
Le 26 avril 2007, le Comité consultatif national d'éthique (CCNE) français a émis un avis s'opposant à l'information des parents des enfants hétérozygotes :
- « [Que] la révélation systématique du statut de porteur sain d'un nouveau-né ne soit pas encouragée, compte tenu de l'absence d'intérêt direct pour l'enfant. »
- « Il s'agit de ne pas transformer un être humain en un être enfermé dans son statut génétique, avec le risque de sacralisation du gène que cela comporte. »
L'information concernant l'hétérozygotie ne devrait être délivrée que sur une demande de l'enfant dépisté et arrivé en âge de procréer mais sachant que ces enfants hétérozygotes et leurs parents sont convoqués pour un test de la sueur, il est difficile de ne pas révéler cette information dès la naissance. De même, les parents d'enfants considérés comme hétérozygotes, sont en droit de s'interroger sur leur propre hétérozygotie notamment dans le cas où ils auraient l'intention d'avoir des enfants dans le cadre d'une autre union.
Dépistage prénatal
Un diagnostic anténatal ou pré-implantatoire est proposé si le conseil génétique détermine qu'un couple a un risque de 25 ou de 50 % d'avoir un enfant malade, ce qui est le cas chez les couples hétérozygotes déjà détectés lors de la naissance d'un enfant malade. Les demandes de couples à faible risque (fratrie de parents d'enfants malades) sont également de plus en plus fréquentes[161],[122].
Chez les couples hétérozygotes, l'analyse de l'ADN du fœtus pour recherche de mutation, par biopsie de villosité choriale peut être réalisé de façon fiable et précoce à 10 semaines d'aménorrhée . Si cette analyse moléculaire est impossible on peut pratiquer une amniocentèse à 18 semaines pour le dosage des isoenzymes de la phosphatase alcaline dans le liquide amniotique. Un taux normal élimine une mucoviscidose alors qu'un taux effondré est évocateur mais non spécifique et nécessite la réalisation d'un caryotype[161],[122].
Une obstruction intestinale fœtale découverte de façon fortuite lors d'une échographie doit faire rechercher la mucoviscidose dont le risque est alors estimé entre 2 et 5 %. Cette anomalie peut être révélée par une hyperéchogénicité ou une dilatation intestinale et elle est secondaire à diverses affections telles qu'un illéus méconial, une atrésie jéjunoiléales, un syndrome de bouchon méconial, un volvulus ou une péritonite méconiale, qui peuvent toutes révéler une mucoviscidose. L'absence de visualisation de la vésicule biliaire au cours de l'échographie fœtale est aussi considérée comme un signe d'appel. Dans ces cas, un diagnostic prénatal est proposé aux parents[161],[162].
Le dépistage anténatal systématique à toutes les femmes enceintes n'a pas été recommandé par le Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE) dans son avis n° 83 du 25 mars 2004[163] qui avait conclu que « le dépistage prénatal est entièrement justifié en cas d'antécédents familiaux ou de connaissance d'hétérozygotie chez un membres du couple, qu'il est à encourager au stade prénuptial ou préconceptionnel pour des familles à risque, qu'il peut trouver une légitimité dans les régions à prévalence importante du gène muté, mais qu'en revanche sa généralisation à l'ensemble d'une population pose des problèmes non seulement éthiques, mais aussi scientifiques, juridiques et économiques. »
Pour les grossesses à haut risque de transmission de la maladie, le diagnostic préimplantatoire est possible chez les couples devant recourir à une procréation médicalement assistée et chez qui la mutation a été identifiée. Ce diagnostic autorisé en France et aux États-Unis mais pas dans tous les pays, consiste à faire l'étude de l'ADN recueilli sur des cellules de l'embryon obtenu après fécondation in vitro. Seuls les embryons non porteurs de la mutation ou hétérozygotes sont transférés.
Conseil génétique
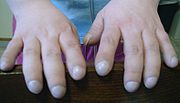
 Mode de transmission autosomique récessive de la mucoviscidose : deux parents porteurs d'une mutation du gène CFTR auront à chaque grossesse, un risque de 1/4 d'avoir un enfant porteur du gène normal, de 1/2 d'avoir un enfant porteur du gène anormal mais non malade, et de 1/4 d'avoir un enfant porteur de deux gènes anormaux et donc malade.
Mode de transmission autosomique récessive de la mucoviscidose : deux parents porteurs d'une mutation du gène CFTR auront à chaque grossesse, un risque de 1/4 d'avoir un enfant porteur du gène normal, de 1/2 d'avoir un enfant porteur du gène anormal mais non malade, et de 1/4 d'avoir un enfant porteur de deux gènes anormaux et donc malade.Le conseil génétique a pour but d'apprécier le risque de récurrence - d'apparition - de la maladie au sein d'une famille à risque. Il découle du mode de transmission de la maladie qui est récessive et non liée au sexe ce qui implique que le risque de récurrence est de 1 sur 4, c'est à dire que si deux parents sont porteurs hétérozygotes d'une mutation pathogène du gène CFTR, ils ont un risque de 25 % d'avoir un enfant homozygote malade. Ils ont également une chance sur 4 d'avoir un enfant sain porteur du gène normal et une chance sur 2 d'avoir un enfant hétérozygote, porteur sain d'une seule allèle anormale et non malade.
Risque pour les membres d'une famille d'un enfant mucoviscidosique
- Frères et sœurs d'un enfant malade
La probabilité d’être hétérozygote pour les frères et sœurs d'un malade est de 2 sur 3. Une recherche de mutation est possible afin de préciser le statut génétique de chaque membre de la fratrie. Cette recherche est importante dans le cadre d’un projet parental; si un porteur sain est détecté, son conjoint peut également bénéficier d'une recherche de mutations afin de déterminer si un risque existe[161].
- Descendant d'un sujet atteint de mucoviscidose
Les femmes sont fertiles, mais il leur est difficile d'avoir un enfant. Les hommes sont stériles à cause de l'atrésie bilatérale des canaux déférents et devront avoir recours aux techniques de procréation médicalement assistée. Chacun des descendants d'un couple dont un des membres est atteint de la mucoviscidose sera porteur d'un gène de la maladie, le risque d'être malade - d'être homozygote - dépendra du statut génétique du conjoint du patient; si celui ci n'est porteur d'aucune mutation, le risque est très faible (mais possible dans le cas d'une mutation rare non détectée); s'il est hétérozygote, le risque sera de 1 sur 2[161]. Le dépistage des mutations du CFTR chez le conjoint dépendra de son histoire familiale et de ses convictions religieuses et personnelles.
Dépistage des porteurs hétérozygotes
Pour que l'enfant présente la maladie, il faut que les deux parents soient porteurs sains hétérozygotes d'une mutation du gène CFTR (ou plus rarement qu'un parent soit atteint de la maladie et l'autre porteur sain hétérozygote). Environ deux millions de personnes sont porteur sain d'une mutation du gène CFTR.
Si les deux parents sont porteurs sains d'une mutation du gène alors l'enfant a une chance sur quatre de porter la maladie, si aucun des deux parents ou un seul est porteur d'une mutation du gène CFTR, il n'y a aucune chance que leurs enfants à naître ait la maladie. Le dépistage d'un porteur sain ne peut se faire que par analyse de son ADN à l'aide de test moléculaire puisqu'il ne présente pas de symptôme. Pour déterminer si les parents d'un couple sont porteurs sains, notamment à l'occasion d'une grossesse, un dépistage de la femme peut être proposé (car c'est elle qui consulte le plus souvent et en cas de test positif), un test peut être fait par la suite sur le père.
Toutefois, cette méthode ne permet pas l'éradication de la maladie. En effet, même si un test prénatal est effectué sur les deux parents, et que l'un de ces tests est négatif, cela ne garantit pas que l'enfant n'aura pas la maladie puisque l'analyse ADN du gène CFTR peut donner de faux négatifs, toutes les mutations du gène n'étant pas connues (la principale mutation ΔF508 représentant moins de 70 % des cas)[163].
Dépistage lors du don de sperme
En France, les généticiens proposent de ne pas dépister systématiquement les donneurs hétérozygotes pour rester dans la même situation qu'en procréation naturelle où le dépistage systématique n'est pas justifié[164].
Évolution et pronostic
L'espérance de vie des patients dépend des possibilités d'accès aux soins et augmente avec l'amélioration de la prise en charge, notamment nutritionnelle et respiratoire. L'espérance de vie des patients est comparable dans les différents pays développés, mais non dans les pays en voie de développement. En l'absence de traitement, la médiane de survie est de 3 à 5 ans[111].
Au Canada, en 1959, l'espérance de vie à la naissance[8] était de 4 ans [165]; en 1977 la vie médiane[166] — qui est l’âge que devrait dépasser la moitié de la population — est de 22,8 ans ; en 1987 elle est de 28,8 ans, en 1997 de 33 ans et en 2002 de 37 ans[167].
En France, les données 2005 du Registre Français de la Mucoviscidose[9], reposant sur l'étude d'environ 80% des patients français, indiquent pour la période 2003-2005 un âge moyen de décès de 24 ans, une espérance de vie à la naissance de 47 ans et une vie médiane de 46,7 ans.
Une étude internationale a retrouvé que l'âge médian au décès était passé de 8 ans en 1974 à 21 ans en 1994[168]. Aux États-Unis, pour les enfants nés en 2006, l'espérance de vie est maintenant de 36,8 années, soit 5 années de plus par rapport au chiffre de 2002[169].
La mucoviscidose touche de façon égale les hommes et les femmes mais pour des raisons encore inexpliquées, l'atteinte est souvent plus grave chez les femmes, avec une infection par Pseudomonas aeruginosa plus précoce, une atteinte pulmonaire et des troubles nutritionnels plus sévères et une espérance de vie plus courte que pour les hommes[99],[100],[101].
Prise en charge et traitement de la maladie
Les progrès de la prise en charge de la mucoviscidose ont permis d'améliorer la qualité et l'espérance de vie des patients ; ainsi en France, l'espérance de vie à la naissance est passée de 7 ans en 1965 à 47 ans en 2005[9].
Les principaux objectifs de la prise en charge sont l'éducation du patient et de sa famille; la prévention, la détection et le traitement précoce des complications broncho-pulmonaires, digestives, rénales, ORL et osseuses; le maintien d'une fonction respiratoire et d'un état nutritionnel optimal, un soutien psychologique régulier et une amélioration globale de la qualité de vie[170].
Aucun traitement curatif, médicamenteux ou thérapie génique, n'est actuellement disponible. Le traitement proposé est symptomatique, destiné à soulager les symptômes de la maladie, et nécessite une prise en charge multi-disciplinaire avec une bonne coordination entre divers spécialistes tels que le pédiatre, le kinésithérapeute, le diététicien ou le psychologue. Il consiste essentiellement en une prise en charge de l'atteinte respiratoire par un drainage bronchique et des antibiotiques, et de l'atteinte digestive et nutritionnelle par une supplémentation en extraits pancréatiques, vitamines et calories[93].
La prise en charge particulièrement lourde et astreignante dans la mucoviscidose entraine pour le patient des contraintes importantes qui retentissent sur sa qualité de vie et sur celle de sa famille et qui favorisent la mauvaise observance thérapeutique, pouvant accélérer l'évolution délétère de la maladie[171].
Traitement palliatif symptomatique
Voies respiratoires
Le traitement palliatif de l'atteinte des voies respiratoires à deux objectifs principaux : l'aide à la clairance muco-ciliaire et le contrôle de l’infection. Pour cela on a recours à la kinésithérapie respiratoire, à des antibiothérapies, à des traitements par bronchodilatateurs, anti-inflammatoires ou fluidifiants mucolytiques. Dans les cas les plus sévères, une greffe pulmonaire ou cœur-poumon peut être envisagée.
Kinésithérapie
Les bronches sont souvent obstruées par une stase de mucus et sujettes à des infections broncho-pulmonaires permanentes. La kinésithérapie a pour objectif de mobiliser et d'évacuer les sécrétions bronchiques, apportant un effet bénéfique immédiat et limitant possiblement à long terme les effets des médiateurs pro-inflammatoires contenu dans les sécrétions muco-purulentes. Elle permet également un entretien global de la mobilité articulaire et du travail musculaire, et permet ainsi le maintien d'une adaptation optimale à l'effort[119].
Dans un premier temps, le kinésithérapeute pratique des techniques d'accélération de flux respiratoire. Il rééduque aussi la toux pour qu'elle devienne plus efficace. Ensuite, le patient va apprendre les techniques de drainage postural et de toilette bronchique qu'il faut pratiquer plusieurs fois par jour. Ce drainage bronchique peut-être aidé par l'utilisation de fluidifiants ou d'aérosol qui augmentent l'hydratation des mucosités et permet leur meilleure mobilisation. La kinésithérapie est particulièrement astreignante pour les patients.
Le kinésithérapeute utilise les techniques de kinésithérapie respiratoire conventionnelle comme le drainage de posture, la percussion et la vibration et les techniques plus récentes utilisant le contrôle du flux expiratoire, la toux contrôlée et l'aide instrumentale. Il peut s'aider de techniques instrumentales telles que [119]:
- l’aérosolthérapie de médicaments (bêta-2-mimétiques, corticoïdes, rhDnase) ou de solutés hypo ou hypertoniques améliorant l’humidification des expectorations,
- la spirométrie incitative,
- le « Threshold inspiratoire » dont le but est d'améliorer l’endurance et la force des muscles inspiratoires,
- l’aspiration des fosses nasales,
- les « PEP systèmes » (Pression Expiratoire Positive) qui entraîne une augmentation du volume de l’expectoration,
- les vibrations mécaniques, externes qui augmentent le volume de l’expectoration et endobronchiques qui pourrait avoir aussi des effets bénéfiques.
Chez un nourrisson dépisté, la kinésithérapie respiratoire quotidienne doit être débutée dès l'apparition d'un symptôme respiratoire à raison d'une séance quotidienne en état stable et de deux séances quotidiennes en période d'exacerbation. L'efficacité préventive de la kinésithérapie respiratoire chez le nourrisson dépisté n'a pas été démontré à ce jour[119].
Antibiothérapie
 Pseudomonas aeruginosa colonise les poumons des personnes atteintes de mucoviscidose sous la forme de biofilm, qui diminue la réponse immunitaire des patients et confère à la bactérie une grande résistance aux antibiotiques[172].
Pseudomonas aeruginosa colonise les poumons des personnes atteintes de mucoviscidose sous la forme de biofilm, qui diminue la réponse immunitaire des patients et confère à la bactérie une grande résistance aux antibiotiques[172]. Staphylococcus aureus au microscope électronique à balayage
Staphylococcus aureus au microscope électronique à balayageLes infections et surinfections bronchiques aiguës ou chroniques qui vont détériorer progressivement la fonction respiratoire, sont traitées par une antibiothérapie déterminée par les examens bactériologiques. La mise en culture des crachats (examen cyto-bactériologique des crachats) ou de prélèvements de sang (hémoculture) permet de déterminer le germe principal en cause, d'évaluer l'importance de la colonisation, et de savoir quels antibiotiques seront efficaces. Les germes principaux (Staphylococcus aureus, Pseudomonas aeruginosa & Burkholderia cepacia) développent habituellement et rapidement des résistances aux antibiotiques. Aussi, les posologies préconisées restent encore mal définies et sont souvent supérieures à celles préconisées dans l’AMM des produits. Il n'est donc pas rare que l'on utilise des doubles antibiothérapies par voie intraveineuse pouvant être accompagnées d'une antibiothérapie inhalée d'entretien. Les aérosols d'antibiotiques doivent être effectués après désencombrement et administration de bêta-2-mimétiques et de rhDNase[119].
Sont généralement utilisés :
- Les bêta-lactamines comme les pénicillines et les céphalosporines, parce que ce sont des antibiotiques à large spectre d'action et souvent bien tolérés même si la prévalence de l'allergie aux bêta-lactamines est plus importante que dans la population générale - de l'ordre de 6 à 22 % des patients - et que leur toxicité hépatique doit être controlée après chaque cure[119]. Parmi les antibiotiques utiles de cette classe, on peut citer la ceftazidime ou le méropénem...
- Les aminosides par voie intraveineuse en association à d'autres antibiotiques contre le Pseudomonas[173], voir la tobramycine utilisée en nébulisation[174]. Leur ototoxicité doit être controlée par un audiogramme annuel, leur néphrotoxicité par des dosages du taux résiduel et par un contrôle de la créatininémie à chaque cure[119].
- Les fluoroquinolones sur le Pseudomonas, même chez les enfants[175] (risque majorée d’arthropathie et de tendinopathie[170]),
- La fosfomycine sur le Pseudomonas multirésistant[176],
- L'acide fucidique sur les Staphyloccoques[177],
- etc.
Concernant la stratégie thérapeutique à adopter vis à vis des principales infections, la Société française de pédiatrie donnait en 2002 les recommandations suivantes[119] :
- Dans l'infection à Staphylococcus aureus, elle ne recommandait pas de traitement prophylactique primaire chez le nourrisson et l’enfant. En première intention elle recommandait, dans les exacerbations à Staphylococcus aureus sensible à la méticilline (SASM), une bêta-lactamine per os, associée ou non à l’acide fusidique, pendant au moins 14 jours. Et dans l'exacerbation à Staphylococcus aureus résistant à la méticilline (SARM), une bithérapie pristinamycine-rifampicine. Elle ne donnait pas de recommandation, devant l'absence de consensus, pour l'antibioprophylaxie secondaire (ou traitement d'entretien).
- Dans l'infection à Pseudomonas aeruginosa (PA) elle recommandait en cas de primocolonisation l'association de deux antibiotiques bactéricides par voie intraveineuse pendant 14 à 21 jours (bêta-lactamine + aminoside), suivis ou non d'aérosols de colistine pendant 3 à 6 mois. Au stade de l'infection chronique, elle recommandait de traiter les exacerbations par une bithérapie afin d'éviter l'émergence de souches résistantes : association d’une bêta-lactamine et de tobramycine pendant au moins 14 jours, ou dans le cas de souches multirésistantes, une trithérapie non validée associant à la bithérapie la ciprofloxacine per os ou la colistine par voie IV. Un traitement d'entretien systématique programmé, par cure d'antibiotiques inhalés de 28 jours ou cures systématiques IV trimestrielles, si possible réalisé à domicile, pouvait être envisagé sans qu'aucune règle ne soit encore établie.
Mucolytiques
Des médicaments mucolytiques (expectorants ou fluidifiants bronchiques) sont utilisables pour rendre le mucus moins visqueux et faciliter son évacuation.
La rhDNase, ou dornase alfa (Pulmozyme®) est une désoxyribonucléase recombinante humaine, similaire à l'enzyme humaine qui hydrolyse l'ADN extracellulaire[178]. Elle agit en 30 minutes en clivant l'ADN des composants du mucus, ce qui en diminue la viscosité et ce qui facilite la clairance liée à la toux. Son administration améliore la fonction respiratoire et diminue le nombre d'exacerbations nécessitant une antibiothérapie. Son efficacité clinique est inconstante et disparaît rapidement après l'arrêt du traitement. Elle est indiquée chez les patients de plus de 5 ans ayant une CVF supérieure ou égale à 40 % de la théorique. Elle s'administre à raison de 1 à 2 nébulisations par jour qui doivent être précédées d'un drainage bronchique proximal et suivies d'une séance de kinésithérapie respiratoire[119].
Bronchodilatateurs
Il n'y a pas assez de preuves de l'intérêt des bronchodilatateurs dans la mucoviscidose pour recommander leur prescription systématique. Les anticholinergiques ne sont pas recommandés car ils n'ont pas démontrer d'efficacité et auraient même une action délétère. Les bêta-2-mimétiques peuvent être utilisés lors des périodes d'exacerbations, au long cours lors des périodes de stabilité et avant le début des séances de kinésithérapie[119].
Corticothérapie
La corticothérapie par voie orale est indiquée en cas d'absence d'amélioration au 14e jour d'une cure d'antibiothérapie prescrite dans le cadre d'une exacerbation. Elle doit être de courte durée, ne dépassant pas deux semaines, en raison des effets délétères sur l'équilibre glycémique et la croissance ainsi que du risque potentiel de favoriser l’émergence d’une colonisation à Pseudomonas aeruginosa[119].
La corticothérapie inhalée n'est pas recommandée en prescription systématique en l'absence d'études suffisantes. Elle est indiquée en présence d'un asthme associé[119].
Oxygénothérapie
Au stade de l'insuffisance respiratoire chronique, une oxygénothérapie (transitoire, de déambulation ou de longue durée) ou une ventilation non invasive peuvent être nécessaires[170].
Greffe pulmonaire
Les patients atteints de mucoviscidose étaient considérés initialement comme de mauvais candidats à la greffe pulmonaire notamment à cause de la surinfection bactérienne chronique des voies aériennes et de leur état nutritionnel précaire. La première transplantation cœur-poumon chez un patient mucoviscidosique est réalisée en 1984 et entre cette date et 2003 plus de 700 transplantations pulmonaires ont été réalisées dans cette indication[179],[79]. En 1999 un comité d'experts publie une conférence de consensus sur la transplantation pulmonaire chez les personnes atteintes de mucoviscidose et conclut que chez le patient atteint de mucoviscidose présentant une insuffisance respiratoire avancée, la transplantation pulmonaire est une option thérapeutique valable[180].
Lorsqu'il y a atteinte sévère de la voie respiratoire, marquée cliniquement par une insuffisance respiratoire avancée, la greffe de poumon (voire cœur/poumon dans certains cas) permet de restaurer une fonction respiratoire correcte et de prolonger la vie du patient mais ne permet pas de guérir la maladie.
La technique de référence est la transplantation bi-pulmonaire et ses résultats et complications sont comparables à ceux observés dans d'autres indications. La plupart des patients reçoivent par la suite une triple thérapie immunosuppressive à vie. À court terme la principale complication est l'infection du greffon, à moyen et long terme c'est le rejet chronique du greffon pulmonaire qui se manifeste par une bronchiolite oblitérante. La survie après greffe pulmonaire est d'environ 70 % à 1 ans, 45 % à 5 ans et 15 % à 10 ans, ces chiffres s'améliorant lentement au fil des années[179].
Une équipe de Los Angeles réalise avec de bon résultats des greffes de deux lobes pulmonaires provenant de donneurs vivants issus de la même famille ou non apparentés[179].
Système oto-rhino-laryngologique
L'atteinte ORL est presque constante dans la mucoviscidose avec notamment sinusite chronique et polypose nasale. La thérapeutique de ces troubles ne fait pas l'objet d'un consensus. La chirurgie généralement proposée face à une polypose, n'évite pas une récidive précoce (à 4 ans en moyenne)[181]. Une chirurgie par voie endoscopique s'est donc développée pour améliorer les suites post-opératoires[182]. Une antibiothérapie par instillation locale, associée à l'endoscopie, diminuerait également les récidives de sinusites et le recours à une chirurgie conventionnelle[183].
Certains antibiotiques, et particulièrement les aminosides utilisés dans la lutte contre la surinfection des voies aériennes par P. aeruginosa, ont une toxicité dose-dépendante sur le rein et l'oreille interne (ototoxicité) et peuvent entrainer des effets secondaires tels que baisse ou perte totale de l'audition. L'apparition d'un trouble de l'audition chez ces patients doit être détectée précocement par une surveillance audiométrique régulière[184].
Prise en charge nutritionnelle et digestive
Les prises en charge de la dénutrition, des manifestations digestives et hépato-biliaires, ainsi que du diabète sont indissociables de la prise en charge des manifestations pulmonaires car chacune de ces atteintes contribue à la morbidité et à la mortalité de la maladie et peut aggraver l’atteinte pulmonaire[171].
Nutrition
La fréquence de la dénutrition est importante dans la mucoviscidose, quel que soit le stade évolutif, et varie selon les études de 15 à 44 % des patients. Il semble qu'un déficit nutritionnel important se rattrape mal par la suite ce qui est en faveur d'une prise en charge précoce de la dénutrition. Dans la mucoviscidose, la dénutrition est due à la fois à la réduction des ingesta (anorexie, inconfort digestif, régimes trop restrictifs, troubles du comportement alimentaire) et à l'augmentation des pertes (insuffisance pancréatique, insuffisance intestinale et perte sudorale et augmentation de la dépense énergétique de repos). Même s'il n'existe pas sur le sujet d’études prospectives contrôlées de grande ampleur, il semble que la dénutrition aggrave fortement la maladie. Pour ces raisons, il faut réaliser une évaluation de l’état nutritionnel dès le dépistage, puis à intervalles réguliers, par évaluation des paramètres cliniques, estimation de la balance énergétique, évaluation des principaux paramètres plasmatiques et étude du statut minéral osseux[171].
Chez le nourrisson dépisté à la naissance, l'allaitement maternel peut être recommandé si la croissance est normale. Chez ceux présentant une insuffisance pancréatique sans cassure des courbes de croissance, les formules lactées classiques semblent tout à fait adaptées, en choisissant celles ayant les plus fortes teneurs en sodium. La diversification alimentaire se fait comme chez l'enfant normal, vers le 5e-6e mois[171].
Au quotidien, les apports énergétiques doivent être légèrement supérieurs aux apports journaliers recommandés (AJR), de l'ordre de 100 à 110 %, ce qui est habituellement suffisant au maintien d'un état nutritionnel normal. Des apports énergétiques supérieurs, de l'ordre de 120 à 150 % des AJR sont souvent inutiles ou difficiles à atteindre par voie orale. Pour maintenir une bonne croissance staturo pondérale, la majorité de patients nécessite moins de 120 % des AJR et seule une minorité a besoin de plus de 120 %, voire plus de 150 %. Pour assurer des apports énergétiques supérieurs aux AJR, on recommande une alimentation riche en calories glucido-lipidiques mais il n'existe pas de consensus sur des valeurs optimales. Il faut orienter précocement les habitudes alimentaires et le goût de l'enfant vers des produits à haute valeur énergétique (produits laitiers, fromages et sucres lents). L'intérêt des compléments hyperénergétiques en l’absence de dénutrition n’est pas démontré. L'intervention d'un diététicien est indispensable et doit être précoce et régulier[171].
Pour prendre en charge l'insuffisance pancréatique exocrine (qui entraine une malabsorption des lipides, mais aussi des glucides et des vitamines liposolubles) on utilise des extraits pancréatiques gastro-résistants qui doivent être pris au début de chaque repas. Les doses recommandées sont exprimées en unités lipase (UL), elles varient selon l'âge du patient et doivent être adaptées à chaque cas. Pour une alimentation normale elles sont chez le nourrisson de 2 000 à 4 000 UL pour 120 ml de lait; chez l'enfant de 1 000 UL/kg/repas ou de 500 UL/kg/collation, sans dépasser 100 000 UL/j ; et chez l'adolescent et adulte de 250 000 UL/jour maximum[171].
De plus, d'autres points sont à surveiller[171] :
- les besoins en eau et en sodium majorés par une sudation importante, surtout pendant les périodes chaudes doivent être compensés.
- des apports complémentaires en vitamines liposolubles A, D et E sont essentiels dès le diagnostic posé et sont apportés sous la forme de complexes polyvitaminiques solubles au double de la posologie habituelle. La vitamine K, rarement carentielle, n'est prescrite que pendant la première année de vie mais peut être nécessaire plus tard lors de traitements antibiotiques prolongés.
- des suppléments en oligo-éléments (fer, zinc, sélénium, magnésium) sont également nécessaires si une carence a été démontrée.
En cas de dégradation de l'état nutritionnel une assistance nutritive est généralement proposée, sans que son bénéfice sur l'état nutritionnel et respiratoire n’est été clairement démontré. Elle repose sur[171] :
- Les suppléments nutritionnels oraux dont l'efficacité n'est pas démontrée et qui ont le désavantage de souvent remplacer les apports oraux habituels ;
- La nutrition entérale (les nutriments sont administrés directement dans l'estomac par une sonde naso-gastrique ou une sonde de gastrostomie) semble d'autant plus efficace qu'elle est commencé tôt. Le mode d'administration est souvent mal toléré ou mal accepté ;
- La nutrition parentérale (les nutriments, sous forme d'une solution lipidique, sont administrés par voie intraveineuse) est lourde et couteuse et nécessite une voie veineuse centrale pour être efficace. Elle peut permettre un gain de poids transitoire mais expose le patient à certains effets secondaires et notamment un risque infectieux. Elle reste d’indication exceptionnelle (pancréatite, post-chirurgicale, refus de nutrition entérale dans un programme de prétransplantation)[170].
Prise en charge des troubles glucidiques
L'aggravation de l'atteinte respiratoire est corrélée à celle de l'atteinte du métabolisme glucidique et l’insulinothérapie améliore les paramètres respiratoires et nutritionnels. De plus avec l’allongement de l’espérance de vie des patients muciviscidosiques, apparaissent les complications microangiopathiques habituellement rencontrées lors du diabète. C'est pourquoi toute hyperglycémie (au stade de diabète voire même d'intolérance au glucose) doit être prise en charge précocement[171].
Le traitement doit privilégier l'insulinothérapie qui est la seule à avoir démontré un effet positif sur le plan nutritionnel, respiratoire, et infectieux. Les antidiabétiques oraux (ADO) sont envisageables dans les formes débutantes et asymptomatiques de diabète ou si l’insulinothérapie est refusée par le patient. Pour les ADO, seuls les insulino-sécrétagogues (sulfamides comme le glimépiride, ou glinides comme le répaglinide ou le natéglinide) sont utilisables. Concernant le choix de l'insuline et du schéma d'insulinothérapie, il est fait en prenant en compte le mode de vie du patient et sa compliance au traitement[171],[118].
Prise en charge de l'atteinte hépato-biliaire
L’atteinte du foie et des voies biliaires au cours de la mucoviscidose peut modifier le pronostic vital et doit être dépistée dès la naissance par un bilan biologique et une échographie. Environ 15 à 20 % des patients développent des lésions hépato-biliaires et cette fréquence augmente nettement à l’adolescence. Le traitement de l’atteinte hépato-biliaire repose sur l'acide ursodésoxycholique (AUDC) qui augmente la sécrétion biliaire et protège les hépatocytes et doit être introduit précocement. Il faut également porter une attention particulière à la toxicité gastrique et hépatique potentielle de certains médicaments. Les complications de la cirrhose menacent le pronostic vital, et l'hypertension portale (HTP) doit être controlée par des techniques endoscopiques, des shunts radiologiques ou chirurgicaux voir la transplantation hépatique[171].
Autres thérapeutiques et hygiène de vie
Toutes les vaccinations du calendrier vaccinal en vigueur (diphtérie, tétanos, polio, coqueluche acellulaire, Haemophilus influenzae type B, pneumococcique, hépatite B, rougeole-oreillons-rubéole) sont recommandées, ainsi que les vaccins contre l’hépatite A et la grippe[170].
Les malades de la mucoviscidose doivent avoir une bonne hydratation et une supplémentation en sel lors des périodes d'efforts et de forte chaleur[185],[186]. La canicule d'août 2003 en France a vu l'augmentation du nombre de cas de déshydratation dans la population des patients mucoviscidosiques[187].
La pratique d’une activité physique ou sportive régulière et adaptée aux capacités respiratoires est fortement conseillée. Un pic VO2 élevé serait associé à une meilleure survie[188]. Une activité physique régulière, par le biais notamment de programme de réhabilitation respiratoire permettrait d'atténuer l'aggravation progressive de l'atteinte de la fonction pulmonaire[189],[190],[191], d'améliorer l'aptitude physique aérobie [190], d'apporter un bien être psychologique[189] et d'augmenter la qualité de vie et la force musculaire des patients[192]. Il n'y a pas encore de standardisation des programmes d'entrainement sportif chez les mucoviscidosiques et des études supplémentaires doivent être menées, afin de valider plus solidement les effets bénéfiques du sport sur ces patients et de mieux déterminer la nature des entrainements et notamment la répartition des exercices aérobie et en résistance[193].
Afin de maintenir un environnement respiratoire de bonne qualité il faut éviter le tabagisme actif ou passif, réduire les risques allergéniques et privilégier un mode de garde individuel plutôt que collectif[170].
Il faut de plus favoriser, par l'élaboration d'un projet individualisé, l'intégration scolaire puis professionnelle. L'intervention de l’assistante sociale et du psychologue doit permettre de faire face aux différentes difficultés[170].
Fertilité chez les femmes
Les troubles du cycle sont fréquents en cas d'atteinte sévère en rapport avec les troubles de la nutrition de la maladie plus que par la maladie elle-même.
Bien que la glaire cervicale soit plus épaisse chez ces femmes leur fertilité semble peu touchée. Une contraception orale est possible malgré une diminution de l'absorption des stéroïdes sexuels qui sont liposolubles chez ces patientes dont l'absorption des graisses est perturbée[194].
Des moyens contraceptifs supplémentaires sont souhaitables lors des traitements antibiotiques par voie veineuse.
Grossesse chez une personne atteinte de mucoviscidose
La première grossesse allant jusqu'à son terme chez une femme atteinte de mucoviscidose fut décrite en 1960, à une époque où la médiane de survie n'était que de 10 ans. La patiente décéda six semaines après son accouchement et les médecins conclurent que la maladie était sérieusement aggravé par la grossesse[195]. Depuis des études ont attesté qu'une grossesse pouvait être envisagée sans trop de risque chez les femmes ayant une bonne fonction pulmonaire[196],[197],[198].
Avec l'amélioration de la qualité et de l'espérance de vie des malades, les grossesses sont de plus en plus nombreuses. En 1992, 4 % des femmes mucoviscidosiques aux États-Unis sont enceintes. La grossesse devrait être planifiée et une recherche des mutations les plus fréquentes doit être faite chez le futur père. Le diagnostic par prélèvement du chorion est fait si le père est porteur d'une mutation puisque le fœtus a un risque sur deux d'être atteint dans ce cas.
En cas d'atteinte sévère, la femme doit être avertie d'un décès possible au cours de la grossesse ou dans le post partum. L'anesthésie générale est compliquée par l'atteinte des poumons. Néanmoins les données disponibles sur les risques exacts de la grossesse chez une femme mucoviscidosique sont faibles. Une réduction de 60 % du volume expiratoire forcé devrait être une contre-indication absolue à une grossesse.
Bien que des grossesses se sont déroulées sans complication après une transplantation cœur-poumon[199], celles-ci sont déconseillées en raison d'une augmentation du rejet du greffon et en raison des risques tératogènes des médicaments anti-rejet.
Organisation des soins et prise en charge sociale
Coordinations des soins
La France a organisé ses soins aux patients malades de mucoviscidose autour de structures dédiées, les centres de ressources et de compétence de la mucoviscidose. Créés par la circulaire n°502 du 22 octobre 2001[200], ces centres sont développés dans des structures hospitalières. Leur rôles est de coordonner les soins délivrés aux malades, de former les professionnels de santé, d'organiser les dépistages, les annonces de diagnostiques, de soutenir la recherche sur la maladie, et d'évaluer tout ce qui tourne autour de la maladie et sa prise en charge. Son travail se développe avec la participation active d'associations de malades, des familles et des patients.
Associations de lutte contre la maladie
Les associations de malades ont pour mission d'encourager la recherche, d'aider au quotidien les malades et leur famille et de former les professionnels de santé. En France, les associations tel que Vaincre la mucoviscidose (organisatrice, en particulier, des virades de l'espoir) se propose d'améliorer la qualité des soins et la qualité de vie des malades et de leurs familles[201]. Plus récemment, l'association Grégory Lemarchal, du nom d'un chanteur vainqueur de la Star Academy ou même le téléthon ont donné un élan médiatique à la maladie.
En plus de son soutien à la recherche et au développement de médicaments contre la mucoviscidose, la Cystic Fibrosis Foundation américaine contribue à financer un réseau national de plus de 115 centres de soins spécialisés consacrés à traiter les gens atteints de mucoviscidose[202],[203].
De nombreux pays où la maladie sévit ont une association d'aide aux malades[204].
Prise en charge financière
En France, la mucoviscidose est une maladie ouvrant droit à la prise en charge en affection de longue durée depuis 1987 (ALD n°18)[170]. L'organisme de sécurité sociale estime le cout annuel des soins remboursés de chaque patient mucoviscidosique à 21 500 euro en moyenne, soit plus de 110 millions d’euros pour l’ensemble des patients. En 2004, concernant les postes de dépense, les médicaments arrivent les premiers avec 8 000 euros (37%), devant l’hospitalisation avec 7 300 euros (34%), les dispositifs médicaux (aérosols, oxygénothérapie, perfusion et certains nutriments) avec 2 300 euros (11%), la kinésithérapie avec 2 200 euros (10%) et enfin les soins infirmiers avec 900 euros. Les coûts de prise en charge varient selon le stade de la maladie. Ainsi la moitié des malades ont une dépense annuelle inférieure à 12 356 euros ce qui ne représente que 10,8% des dépenses alors que pour 10% des patients, les coûts s'élève à plus de 51 200 euro[205].
En 1996, en Californie, une petite étude sur une population de souscripteurs à une assurance médicale, estime le cout des soins aux patients mucoviscidosiques en moyenne à 13 300$ avec des écarts allant de 6 200$ pour des patients peu atteints et jusqu'à 43 300$ pour les formes plus sévères de la maladie[206].
L'étude des variations des couts des soins médicaux ont permis de retrouver des facteurs prédisposant aux soins plus onéreux. Une étude sur des patients de l'État d'Alberta retrouve l'âge, le sexe, l'utilisation de rhDNAse et les infections à Pseudomonas et Burkholderia comme des déterminants majorant les couts de traitement[207].
Organisation académique
En France ont été publiées en 2002, sous l'égide de la Société française de pédiatrie (SFP) et de la Haute Autorité de santé (HAS), des recommandations à l'usage des professionnels de la santé, portant sur les poumons et l'infectiologie[119] d'une part, et sur l'aspect nutritionnel, gastro-entérologique et métabolique[171] d'autre part.
L’European Cystic Fibrosis Society est composé de médecins impliqués dans la prise en charge de la maladie et a été formée depuis la fin des années 1960[208]. Il édite une revue scientifique entièrement consacrée à ce sujet, The Journal of cystic fibrosis[209]. Il publie également des recommandations sur les différents aspects de la prise en charge de la maladie[210].
Voies de recherches
Modèle animal
Un des objectifs essentiels pour faire avancer la recherche sur la mucoviscidose est la création d'un modèle animal permettant une meilleure compréhension des mécanismes physiopathologiques de la maladie, une identification des gènes et des facteurs environnementaux modifiant la sévérité du phénotype, l'expérimentation de nouvelles stratégies pharmacologiques modifiant la sévérité de la maladie et l'étude de protocoles de thérapie génique corrigeant la défaillance du transport ionique[211]..
En 1992, trois ans à peine après la découverte du gène CFTR, la première souris « knockout » pour CFTR est créé par ciblage génétique ; elle présente à l'état homozygote de nombreux symptômes communs avec les jeunes patients humains, tels une stérilité, un iléus méconial, des altérations des glandes muqueuses et séreuses et des obstructions des structures glandulaires, entrainant la mort par obstruction intestinale avant 40 jours de vie[212]. Par la suite ont été décrits de nombreux autres modèles murins modifiés génétiquement de façon à entrainer soit une absence complète d’expression de CFTR, soit l'expression d'un CFTR muté[211]. Ces modèles animaux présentaient des similitudes, mais aussi des différences avec la maladie humaine : ainsi la plupart développaient des anomalies du transport ionique et des symptômes digestifs similaires à l'homme, mais ne présentaient pas les lésions pulmonaires caractéristiques retrouvées chez les patients humains, alors que l'atteinte digestive leur était en général fatale[211]. Afin de contourner cette absence de phénotypie respiratoire, des modèles de souris différemment sensibles à certains pathogènes respiratoires ont été crée, permettant de mettre en évidence une susceptibilité augmentée pour ces pathogènes chez les souris déficientes en CFTR[213]. Cependant ces modèles étaient peu satisfaisants car ils ne reproduisaient pas les manifestations de la maladie pulmonaire chez l’humain[214].
Après 15 ans de recherche d'un modèle animal permettant d'étudier la physiopathologie de l'atteinte pulmonaire de la mucoviscidose, Mall et coll. décrivent en 2004 un nouveau modèle murin présentant un phénotype respiratoire très proche de celui retrouvé dans la mucoviscidose chez l’homme. Pour créer ce modèle ils se sont basés sur la surexpression du canal au Na+ ENaC contrôlé par CFTR, et non pas sur la déficience du CFTR[215]. Leur modèle sous entend que c'est l'altération de la clearance muco-ciliaire qui entraine l'inflammation bronchique, et que celle-ci précède l'infection chronique ce qui permet d'envisager de nouvelles voies de recherches thérapeutiques en cherchant à corriger le défaut de régulation des flux de Sodium trans-épithéliaux[214].
Traitement curatif
Thérapie génique
La mucoviscidose étant une maladie monogénétique, c'est-à-dire ne concernant qu'un seul gène, c'est naturellement que de grands espoirs de guérison sont nés avec l'apparition du concept de la thérapie génique. Des analyses de la quantité d'ARN messager (ARNm) dans les cellules saines ont révélés un nombre extrêmement faible d'ARNm codant CFTR, entre deux et trois copies par cellule. En théorie, même avec un taux de transfert très faible, il serait possible de restaurer une fonction de sécrétion normale dans les cellules pulmonaires en apportant une ou deux copies du gène sain intégré dans un vecteur. La fonction restaurée, le mucus devrait se fluidifier et permettre une clairance muco-cilliaire satisfaisante. En effet, les infections des voies pulmonaires par des pathogènes et l'inflammation qui s'ensuit est l'une des cause de la perte de la fonction respiratoire.
Le vecteur choisi a été l'adénovirus car il infecte de manière sélective les cellules pulmonaires. On réalise une infusion de virus transformés directement dans les bronches du patient. Malgré un principe relativement simple, la thérapie génique bute sur plusieurs points[216],[217]. Le système immunitaire du patient lutte contre le vecteur adénoviral et le détruit. En outre les cellules ayant intégré le transgène expriment des protéines du virus et sont donc identifiées puis éliminées par le système immunitaire cellulaire. De par la présence d'un mucus bronchique épais, la pénétration de l'adénovirus dans les cellules cibles est considérablement ralentie. Par ailleurs, le transgène n'est pas intégré de manière définitive dans les cellules cible et il est assez rapidement éliminé.
Thérapie protéique
La connaissance des mécanismes physiopathologiques dans la mucoviscidose laisse la possibilité d'un traitement pharmacologique visant à intervenir pour pallier leur défaillance.
Activation de la protéine CFTR
De nombreuses molécules semblent pouvoir intervenir sur la maturation de la protéine ΔF508-CFTR et empêcher son blocage prématuré par l'appareil de Golgi, augmentant ainsi le nombre de molécule transmembranaire CFTR atteignant la paroi apicale des cellules ciliées et fournissant un travail de transfert du chlore.
Parmi elles, il y a eu des essais avec le butyrate et ses composés[218] ; in vitro, de hautes concentrations de glycérol ou de N-oxyde triméthylamine, le myoinositol et la taurine[219] peuvent partiellement neutraliser le défaut de transfert de la protéine ΔF508 CFTR vers la surface membranaire.
Le Miglustat[220] est testé depuis septembre 2007 dans un essai clinique de niveau II comme inhibiteur de l'enzyme de glycosylation de la protéine F508del-CFTR. La glycosylation entraînant une dégradation accélérée de la protéine par le réticulum endoplasmique, son inhibition permet de laisser le CFTR même muté, prendre ses fonctions sur la membrane apicale de la cellule ciliée[221].
Activation de voies alternatives de sécrétion
Devant l'échec de la thérapie génique, d'autres stratégies thérapeutiques sont envisagées ; l'une d'entre elles consiste en l'activation de voies de sécrétion saines.
Le canal chlorure CFTR n'est pas le seul canal chlorure apical présent dans les épithéliums. L'idée sous-jacente de cette stratégie thérapeutique est d'activer à l'aide de médicaments spécifiques des canaux déjà présents dans la membrane apicale. Les études sont rendues difficiles de par la difficulté que représente l'obtention d'un épithélium de poumon fonctionnel. C'est la raison pour laquelle la grande majorité des travaux sont faits sur l'épithélium colique, un autre épithélium qui présente une sécrétion de chlorure de sodium dépendante de l'AMPc. À l'aide de cet outil, plusieurs voies alternatives ont été décrites par les chercheurs. Parmi celles-ci, on peut citer la tentative d'activer la sécrétion de chlorure de calcium dépendant du calcium en activant les canaux purinergiques P2X à l'aide d'ATP. Le traitement testé a donc consisté en l'injection, à l'aide d'un inhalateur, d'un agoniste des récepteurs purinergiques P2X. La liaison de l'agoniste sur ces récepteurs provoque une augmentation de calcium intracellulaire, calcium qui va activer la sécrétion de chlorure de calcium en activant un canal chlorure dépendant du calcium apical. À la différence de la sécrétion de calcium passant par CFTR, la sécrétion ainsi induite n'est pas soutenue mais transitoire, des phénomènes de rétroinhibition stoppant assez rapidement l'activation de cette voie.
Personnalités atteintes de la mucoviscidose
Nom Naissance et décès Notes Référence Lisa Bentley (1968 -) Ironman triathlète canadienne. [222] - (en) Christopher Davies (1978 -) Ancien joueur de cricket des Southern Redbacks et actuel coach du Melbourne Cricket Club. [223] - (en) Bob Flanagan (1952 – 1996) Écrivain, poète, artiste de performance et comique américain. [224] - (en) Nolan Gottlieb (1982 -) Ancien joueur de basket-ball NCAA de l'Anderson University et actuel coach assistant du même club. [225] - (en) Grégory Lemarchal (1983 - 2007) Chanteur français, vainqueur de la quatrième saison de la Star Academy. [226] Alice Martineau (1972 - 2003) Chanteuse et modèle britannique. [227] - (en) Andrew Simmons (1984 -) Catcheur britannique. [228] - (en) Bill Williams (1960 - 1998) Développeur informatique. [229] - (en) Certains scientifiques Polonais, se basant sur des faits de la vie de Frédéric Chopin, soupçonnent ce dernier d'être mort d'une mucoviscidose et non d'une tuberculose comme la thèse actuelle le laisse penser[230]. Cependant, le gouvernement polonais a refusé l'analyse ADN du cœur du pianiste actuellement conservé dans une église de Varsovie.
Sources et références de l'article
Sources
- (en) Samuel M Moskowitz, Ronald L Gibson, Darci L Sternen, Edith Cheng, Garry R Cutting. CFTR-Related Disorders in GeneTests: Medical Genetics Information Resource. Copyright, University of Washington, Seattle. 1993-2007.
- Pour le chapitre « Historique »
- (en) Jim Littlewood, « The history of the development of cystic fibrosis care. Perspective of a general paediatrician at a provincial teaching hospital. » sur 2002, The UK CFTrust, London, UK.
- (en) Quinton P. M., « Physiological basis of cystic fibrosis: a historical perspective », dans Physiological reviews (ISSN 0031-9333), no 1, 1999, 79, p. 3-22 résumé texte intégral.
Références
- ↑ a , b , c et d (en) Cystic Fibrosis Mutation DataBase Statistics sur http://www.genet.sickkids.on.ca. Consulté le 12 juin 2008
- ↑ a , b , c et d Emmanuelle Girodon-Boulandet, Catherine Costa, « Génétique de la mucoviscidose », dans Médecine thérapeutique / Pédiatrie, vol. 8, no 3, mai-juin 2005, p. 126-34 [texte intégral (page consultée le 27 mai 2008)]
- ↑ a et b (en) Corey M, Farewell V. Determinants of mortality from cystic fibrosis in Canada, 1970-1989. American Journal Epidemiology 1996 May 15;143(10):1007-17.
- ↑ a et b Plan national maladie rare 2005-2006. Consulté le 27 mai 2008
- ↑ a et b (en) About Cystic Fibrosis sur www.cff.org. Consulté le 28 mai 2008
- ↑ Mucoviscidose est un terme inspiré des travaux du pédiatre américain Farber (mucoviscidosis, en 1943), qui conçut l'hypothèse d'un mécanisme pathologique commun, entrainant des sécrétions muqueuses trop visqueuses, pour regrouper en une même entité pathologique systémique, des atteintes poly-organiques souvent associées (digestives, respiratoires, biliaires, sudoripares...).
- ↑ Cette dénomination anglophone de CF ou Cystic Fibrosis (« fibrose kystique ») est relativement réductrice, en se focalisant sur un type particulier d'atteinte, pour un organe en particulier, le pancréas. Cependant, la fibrose kystique du pancréas est une atteinte fréquente, caractéristique de la maladie, historiquement une des premières à avoir alerté les équipes pédiatriques.
- ↑ a et b Définition : l'espérance de vie à la naissance, ou vie moyenne, est une donnée statistique exprimant le nombre moyen d'années que peut espérer vivre un enfant né à une certaine date, si les conditions de mortalité ayant prévalu au cours de cette période demeurent inchangées durant toute sa vie.
- ↑ a , b et c Registre Français de la Mucoviscidose (RFM) : Bilan des données 2005 de l'Observatoire National de la Mucoviscidose (ONM), Vaincre la Mucoviscidose et Institut National d’Etudes Démographiques (INED), Paris, 2007
- ↑ a , b et c (de)Fanconi G, Uehlinger E, Knauer.C. Das coeliakiesyndrom bei angeborener zysticher pankreasfibromatose und bronchiektasien. Wien Med Wschr 1936; 86:753-756.
- ↑ a et b « The child will soon die whose brows tastes salty when kissed. » Ernst Ludwig Rochholz, Almanac of Children’s Songs and Games from Switzerland. Publié en 1857 par J. J. Weber. Titre original : Alemannisches Kinderlied und Kinderspiel aus der SchweizGesammelt (Leipzig 1857). Consulter le livre.
- ↑ a et b « Wehe dem Kind, das beim Kuß auf die Stirn salzig schmekt, es ist verhext und muss bald sterben », en anglais « Woe to the child kissed on the brow who tastes salty, for he is cursed and soon must die. » Welsh MJ and Smith AE. Cystic fibrosis. Sci Am 273: 52–59, dec 1995. PMID: 8525348, ISI.
- ↑ a et b (en) Busch R. On the history of cystic fibrosis. Acta Univ. Carol. 36: 13-15, 1990. PMID: 2130674
- ↑ Alonso y de los Ruyzes de Fonteca, J. Diez Previlegios para Mgeres Preñadas. Henares, Spain: Alcalá de Henares, 1606, p. 212
- ↑ (en) Busch R. On the history of cystic fibrosis. Acta Univ Carol [Med] (Praha). 1990;36(1-4):13-5. PMID 2130674
- ↑ Landsteiner K. Darmverschluss durch eingedicktes Meconium. Pankreatitis. Zentrabl Allg Pathol 1905; 16:903-907. (Cité par Super M. Milestones in cystic fibrosis. Brit Med Bull 1992; 48:717-737.2)
- ↑ (en) Garrod AE, Hurley WH. Congenital familial steatorrhoea. Q J Med 1912; 6:242-258. qjmed
- ↑ a et b (en) Andersen DH, Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child 1938; 56:344-399
- ↑ (en) Blackfan, K. D., and C. D. May. Inspissation of secretion, dilatations of the ducts and acini, atrophy and fibrosis of the pancreas in infants. J. Pediatr. 13: 627-634, 1938.
- ↑ (en) Harper, M. H.. Congenital steatorrhoea due to pancreatic defect. Arch. Dis. Child. 13: 45-56, 1938. Résumé
- ↑ (en) Andersen DH. Cystic fibrosis of the pancreas, vitamin A deficiency and bronchiectasis. J Pediatr 1939; 15:763-771.
- ↑ (en) Andersen, Dorothy H. The Present Diagnosis and Therapy of Cystic Fibrosis of the Pancreas. Proc R Soc Med. 1949 Jan;42(1):25–32. PMID: 18123627
- ↑ a et b (en) Andersen DH. Therapy and prognosis of fibrocystic disease of the pancreas. Pediatrics 1949; 3: 406-417. Résumé, Texte complet
- ↑ (en) Farber S. Pancreatic insufficiency and the celiac syndrome. N Engl J Med 1943; 229:653-682.
- ↑ Quinton P. M., « Physiological basis of cystic fibrosis: a historical perspective », dans Physiological reviews, vol. 79, no 1, 1999, p. 3-22 (ISSN 0031-9333) [résumé, texte intégral (pages consultées le 28 mai 2008.)]
- ↑ (en) Andersen DH, Hodges RC. Celiac syndrome. V. Genetics of cystic fibrosis of the pancreas with a consideration of etiology. Am J Dis Child 1946; 72:62-80.
- ↑ (en) Kessler, W. R. and D. H. Andersen. Heat prostration in fibrocystic disease of the pancreas and other conditions. Pediatrics 8: 648-655, 1951. Résumé
- ↑ (en) Darling RC, diSant'Agnese PA, Perera GA, Andersen DH. Electrolyte abnormalities of the sweat in fibrocystic disease of the pancreas. A J Med Sci 1953; 225:67-70. PMID: 13007698.
- ↑ (en) Di Sant' Agnese PA, Darling RC, Perera GA, et al. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: clinical implications and relationship to the disease. Pediatrics 1953; 12:549–563. PMID: 13111855, Abstract
- ↑ a et b (en) Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilising pilocarpine electrophoresis. Pediatrics 1959; 23:545-549. abstract
- ↑ (en) Shwachman H, Mahmoodian A. Pilocarpine iontophoresis sweat testing : Results of seven years'experience. Fourth International Conference on Cystic Fibrosis of the Pancreas (Mucoviscidosis). Berne/Grindewald 1966, part I Mod Probl Paediatr 1967 ; 10 : 158. PMID:6054626
- ↑ (en) Legrys VA. Sweat testing for the diagnosis of cystic fibrosis : practical considerations. Pediatrics 1996 ; 129 : 892-7. PMID: 8969732
- ↑ (en) Knowles MR, Gatzy JT, Boucher RC. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Eng J Med 1981; 305:1489-1495. Résumé
- ↑ (en) Quinton, P. M. Suggestion of an abnormal anion exchange mechanism in sweat glands of cystic fibrosis patients. Pediatr. Res. 16: 533-537, 1982 PMID: 7110773
- ↑ (en) Quinton PM. Chloride impermeability in cystic fibrosis. Nature 1983; 301:421-422. PMID: 6823316
- ↑ Laurence Nieto, « Le clonage positionnel : exemple du gène de la mucoviscidose » sur www.ipbs.fr
- ↑ (en) Eiberg H, Mohr J, Schmiegelow K, Nielsen LS, Williamson R. Linkage relationships of paraoxonasse (PON) with other genetic markers: indication of cystic fibrosis synteny. Clin. Genet. 28: 265-271, 1985. PMID: 2998653
- ↑ (en) Tsui L, Buchwald M, Barker D, Braman JC, Knowlton R, Schumm JW, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 1985; 230:1054-1057. Résumé
- ↑ (en) Kerem B-S, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M et Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245:1073-1080. Résumé. PMID 2570460.
- ↑ (en) Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zelenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS et Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of the complementary DNA . Science 1989; 245:1066-1073. Erratum dans Science 1989 Sep 29;245(4925):1437. Résumé. PMID 2475911.
- ↑ (en) Rommens JM, Iannuzzi MC, Kerem B-S, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Keneddy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui LC et Collins FS. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245:1059-1065. Résumé. PMID 2772657.
- ↑ (en) Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, et Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253: 202-205, 1991. Résumé
- ↑ (en) Bear CE, Li CH, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M, Riordan JR. Purification and functional reconsititution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68: 809-818, 1992. PMID: 1371239
- ↑ a , b , c , d , e , f , g , h , i et j (en) Jim Littlewood, « The history of the development of cystic fibrosis care. Perspective of a general paediatrician at a provincial teaching hospital. » sur http://www.cysticfibrosismedicine.com, 2002, The UK CFTrust, London, UK. Consulté le 24 juin 2008
- ↑ (en) Andersen DH.(a) Celiac syndrome II. Fecal excretion in congenital pancreatic deficiency at various ages with various diets, with discussion of the optimal diet. Am J Dis Child 1945; 69:221.
- ↑ (en) Kennedy RLJ. Cystic fibrosis of the pancreas. Nebraska M J 1946; 31:493-496.
- ↑ (en) Di Sant'Agnese PEA, Andersen DH. Celiac Syndrome IV Chemotherapy I infections of the respiratory tract associated with cystic fibrosis of the pancreas; observations with penicillin and drugs of the sulphonamide group, with special reference to penicillin aerosol. Am J Dis Child 1946; 72:17-61.
- ↑ (en) Gandevia B, Anderson CM. The effect of bronchodilator aerosol on ventilatory capacity in fibrocystic disease of the pancreas. Arch Dis Child 1959; 34:511-515. PMID: 13826268, Texte intégral
- ↑ Poncher, H Editor. Year Book of Pediatrics, 1951
- ↑ (en) Stoppelman MRH, Shwachman H. Effect of antibiotic therapy on mucoviscidosis: bacteriologic study. New Eng J Med 1954; 251:759-763. PMID: 13214325
- ↑ (en) Shwachman H, Silverman BK, Patterson, Zheutlin LJ. Antibiotics in treatment of pancreatic fibrosis with emphasis on terramycin. JAMA 1952; 149:1101-1108. PMID: 14938116
- ↑ (en) Shwachman H, Kulczycki LL. Long-term study of 105 cystic fibrosis patients. Am J Dis Child 1958; 96:6-15. Texte intégral
- ↑ (en) Bishop HC, Koop CE. Management of meconium ileus, resection, Roux-en-Y anastomosis and ileostomy irrigation with pancreatic enzymes. Ann Surg 1957; 50:835-836. PMID: 13403593, Texte intégral
- ↑ (en) Harris R, Norman AP, Payne WW. The effect of pancreatin therapy on fat absorption and nitrogen retention in children with fibrocystic disease of the pancreas. Arch Dis Child 1955; 30:424-427. Texte intégral
- ↑ (en) Matthews LW, Spector S. Breakthrough in cystic fibrosis. Pediatrics 1961; 27:351. PMID: 13768289
- ↑ (en) Matthews LW, Doershuk CF, Spector S. Mist tent therapy of obstructive pulmonary lesion of cystic fibrosis. Pediatrics 1967; 39:176-185. PMID: 6017955, Texte intégral
- ↑ (en) Bau SK, Aspin N, Wood DE, Levison H. The measurement of fluid deposition in humans following mist tent therapy. Pediatrics 1971; 48:605-612. PMID: 5114748, Texte intégral
- ↑ (en) Chang N, Levison H, Cunningham et al. An evaluation of nightly mist therapy for patients with cystic fibrosis. Am Rev Resp Dis 1973; 107:672-675. PMID: 4697676
- ↑ (en) Alderson PO, Secker-Walker RH, Strominger DB, Markham J, Hill RL. Pulmonary deposition of aerosols in children with cystic fibrosis. J Pediatr 1974; 84:479-484. PMID: 4599607
- ↑ (en) Lawson D. Contribution to a Panel discussion on microbiology and chemotherapy of the respiratory tract in cystic fibrosis. In Proceedings of the 5th International Cystic Fibrosis Conference, Cambridge, September 1969, p225. Ed: Lawson D. London, Cystic Fibrosis Research Trust, 1969.
- ↑ (en) Lawson D. Cystic fibrosis-Assessing the effects of treatment. Arch Dis Child 1972; 47:1-4. PMID: 5018653, Texte intégral
- ↑ (en) Mearns MB. Treatment and prevention of pulmonary complications of cystic fibrosis in infancy and early childhood. Arch Dis Child 1972; 47:5-11. PMID: 5018658, Texte intégral
- ↑ (en) Gracey M. Cystic fibrosis. In: Anderson CM, Burke V, editors. Paediatric Gastroenterology. Oxford, Blackwell, 1975; 329-360
- ↑ (en) Allan JD, Mason A, Moss AD. Nutritional supplementation in treatment of cystic fibrosis of the pancreas. Am J Dis Child 1973; 126:2-26. PMID: 4723167.
- ↑ (en) Kraemer R, Rudeberg A, Hadorn B, et al. Relative underweight in cystic fibrosis and its prognostic value. Acta Paediatr Scand 1978; 67:33-37. PMID: 626067
- ↑ (en) Noblett HR. Treatment of uncomplicated meconium ileus by gastrografin enema. A preliminary report. J Pediatr Surg 1969; 4:190-197. PMID: 5778338
- ↑ (en) Govan JR, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M, et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 1993; 342:15-19. PMID: 7686239
- ↑ (en) Hoiby N. Antibodies against Pseudomonas aeruginosa in serum from normal persons and patients colonised with mucoid or non-mucoid P. aeruginosa: results obtained by crossed immunoelectrophoresis. Acta Pathol Microbiol Immunol Scand 1977; 85:142-148. PMID: 404843.
- ↑ (en) Schiotz PO, Hoiby N, Flensborg EW. Cystic fibrosis in Denmark. In: Warwick WJ. Ed: 100 years of Cystic Fibrosis. Minnesota 1981:141-146.
- ↑ a et b Szaff M, Hoiby N, Flensborg EW. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr Scand 1983; 72; 651-657. PMID: 6637463
- ↑ (en) Jensen T, Pedersen SS, Hoiby N, Flensborg EW. Use of antibiotics in cystic fibrosis. The Danish Approach. In: Pseudomonas aeruginosa infection. Antibiot Chemother. Eds: Hoiby N, Pedersen SS, Doring G, Holder IA. Basel, Karger, 1989; 42:237-246.
- ↑ (en) Crozier DN. Cystic fibrosis: a not so fatal disease. Pediatr Clin North Am 1974; 21:935-948. PMID: 4610494.
- ↑ (en) Norman AP. Cystic Fibrosis and normality. In: Warwick WJ. 1000 Years of Cystic Fibrosis. Minnesota 1981:84-89.
- ↑ (en) Phelan P, Hey E. Cystic fibrosis mortality in England and Wales and in Victoria Australia. Arch Dis Child 1984; 59:71-83. Arch Dis Child
- ↑ (en) Rabin HR, Harley FL, Bryan LE, Elfring GL. Evaluation of high dose tobramycin and ticarcillin treatment protocol in cystic fibrosis based on improved susceptibility criteria and antibiotic pharmacokinetics. In: Perspectives in Cystic Fibrosis. Ed. Sturgess JM. 8th International Cystic Fibrosis Congress, Toronto, Canada 1980:370-375.
- ↑ (en) Conway SP, Miller MG, Ramsden C, Littlewood JM. Intensive treatment of pseudomonas chest infection in cystic fibrosis: a comparison of tobramycin and ticarcillin and netilmicin and ticarcillin. Acta Paediatr Scand 1985; 74:107-113. Acta Paediatrica
- ↑ (en) Hodson ME, Penketh ARL, Batten JC. Aerosol carbenicillin and gentamicin treatment of Pseudomonas aeruginosa in patients with cystic fibrosis. Lancet 1981; ii: 1137-1139. PMID: 6118579
- ↑ (en) Littlewood JM, Miller MG, Ghoneim AT, Ramsden CH. Nebulised colomycin for early pseudomonas colonisation in cystic fibrosis. Lancet 1985; I: 865. PMID: 2858720
- ↑ a et b (fr) P Foucaud , G. Lenoir et J. Navarro, « Mucoviscidose : transplantation pulmonaire » sur http://www.lesjta.com, 1998. Consulté le 26 juin 2008
- ↑ (en) Pasque MK, Cooper JD, Kaiser LR, et al. Improved technique for bilateral lung transplantation: rationale and initial clinical experience. Ann Thorac Surg 1990; 49:785-791. PMID: 2339934,The Annals of Thoracic Surgery
- ↑ (en) Starnes VA, Barr M, Cohen R, et al. Living donor lobar lung transplantation experience: intermediate results. J Thorac Cardiovasc Surg 1996; 112:1284-1291. PMID:8911325,J Thorac Cardiovasc Surg
- ↑ (en) MacDonald A. High moderate or low fat diets for cystic fibrosis? In Cystic Fibrosis: Horizons. Ed: Lawson D. John Wiley & Sons. 1984; 395.
- ↑ (en) Littlewood JM, MacDonald A. Rationale of modern dietary recommendations in cystic fibrosis. J Roy Soc Med 1987; 80 (Suppl 15):s16-s24. PMID: 3116241,Texte complet
- ↑ (en) Davies JC, Geddes DM, Alton EWFW. Gene therapy for cystic fibrosis. J Gene Medicine 2001; 3:409-417. PMID: 11601754
- ↑ (en) Rodgers HC, Knox AJ. Pharmacological treatment of the biochemical defect in cystic fibrosis airways. Eur Resp J 2001: 17:1314-1321. PMID: 11491179, Texte complet
- ↑ (en) Clinical Guidelines for the Physiotherapy Treatment of Cystic Fibrosis, UK CF Trust, Association of Chartered Physiotherapists in Cystic Fibrosis (ACPCF), Recommendations of a Working Group, January 2002.
- ↑ (en) Serre JL, Simon-Bouy B, Mornet E, Jaume-Roig B, Balassopoulou A, Schwartz M, Taillandier A, Boué J, Boué A. Studies of RFLP closely linked to the cystic fibrosis locus throughout Europe lead to new considerations in population genetics. Hum Genet 84:449–454 (1990). PMID: 1969843
- ↑ (en) Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T, Giménez J, Reis A, et al. The origin of the major cystic fibrosis mutation (ΔF508) in European populations. Nat Genet 7:169–175 (1994). PMID: 7920636.
- ↑ (en) Wiuf C. Do ΔF508 heterozygotes have a selective advantage? Genet Res 78:41–47 (2001). PMID: 11556136.
- ↑ (en) Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet 17:502–510 (2001). PMID: 11525833.
- ↑ (en) Morral N, Nunes V, Casals T, Chillón M, Giménez J, Bertranpetit J, Estivill X. Microsatellite haplotypes for cystic fibrosis: mutation frameworks and evolutionary tracers. Hum Mol Genet 2:1015–1022 (1993). PMID: 7689896 .
- ↑ (en) Mateu E. Can a place of origin of the main cystic fibrosis mutations be identified? Am J Hum Genet 2002, 70:257-264. Article complet, PMID: 11713719
- ↑ a , b , c et d (fr) Pr . G. Bellon, « Mucoviscidose » sur http://www.orpha.net, avril 2006. Consulté le 27 mai 2008
- ↑ (en)Racial/Ethnic Differences, sur http://www.lungusa.org, le site de l'American Lung Association.
- ↑ Données épidémiologiques d’après Monaghan et Feldman, Prenat Diagn 1999; 19: 604-9. Cité par Girodon-Boulandet dans Génétique de la mucoviscidose, Médecine thérapeutique / Pédiatrie. Volume 8, Number 3, 126-34, mai-juin 2005.
- ↑ a et b [pdf] Prévalence des maladies rares, une enquête bibliographique, février 2008 sur www.orpha.net
- ↑ Note : pour la mucoviscidose, maladie létale apparaissant à la naissance, la prévalence = incidence à la naissance x (espérance de vie des malades/espérance de vie de la population générale). Voir la référence précédente pour plus de détails.
- ↑ Dommergues M, Aymé S, Janiaud P, Séror V. Diagnostic prénatal: pratiques et enjeux. Paris: Éditions Inserm, 2003. 571 p. Fiche descriptive de l'ouvrage.
- ↑ a et b (en) Demko CA, Byard PJ, Davis PB. Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J Clin Epidemiol. 1995;48:1041-1049. PMID: 7775991
- ↑ a et b (en) O'Connor GT, Quinton HB, Kahn R, et al. Case-mix adjustment for evaluation of mortality in cystic fibrosis. Pediatr Pulmonol. 2002;33:99-105. PMID: 11802245
- ↑ a et b (en) Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B. Gender gap in cystic fibrosis mortality. Am J Epidemiol. 1997;145:794-803. Résumé, Texte complet
- ↑ A. Sangare, I. Sanogo, E. Ebongo, M. Meite, P. Kple Faget, S. Sawadogo, A. Segbena, V. Ambofo, J. Ohoun, G. Assale. Contribution à l'étude des relations entre la drépanocytose et le paludisme. Médecine d'Afrique Noire : 1990, 37 (5). Texte complet.
- ↑ (en) Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science. 1994 Oct 7;266(5182):107-9. PMID 7524148
- ↑ (en) Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ. The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J Physiol. 1995 Jan 15;482 (Pt 2):449-54. PMID 7714835
- ↑ (en) Hogenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, Prestidge CB, Fordtran JS. Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion. Am J Hum Genet. 2000 Dec;67(6):1422–7. Epub 2000 Oct 30. PMID 11055897.
- ↑ (en) Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, Ratcliff R, Evans MJ, Colledge WH. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature. 1998 May 7;393(6680):79–82. PMID 9590693
- ↑ (en) Poolman EM, Galvani AP. Evaluating candidate agents of selective pressure for cystic fibrosis. J R Soc Interface. 2007 Feb 22;4(12):91-8. PMID: 17015291, Texte complet.
- ↑ a et b (en) CFTR: The Gene Associated with Cystic Fibrosis sur http://www.ornl.gov, 12 septembre 2003, Genomics.energy.gov. Consulté le 6 juin 2008
- ↑ a et b (fr) CFTR sur http://www.ncbi.nlm.nih.gov, OMIM (National Center for Biotechnology Information). Consulté le 6 juin 2008
- ↑ (en) CFTR at Genetics Home Reference sur http://ghr.nlm.nih.gov, 30 mai 2008, Genetics Home Reference. Consulté le 6 juin 2008
- ↑ a , b , c , d , e et f (fr) Fanen P, Hasnain A, « Mucoviscidose et Gène CFTR » sur http://atlasgeneticsoncology.org, septembre 2001, Atlas Genet Cytogenet Oncol Haematol. Consulté le 12 juin 2008
- ↑ (en) McKone EF, Emerson SS, Edwards KL, Aitken ML, Effect of genotype on phenotype and mortality in cystic fibrosis/ a retrospective cohort study Lancet. 2003 May 17;361(9370):1671-6. PMID 12767731
- ↑ (en) Ferrari M, Cremonesi L. Genotype-phenotype correlation in cystic fibrosis patients. Ann Biol Clin (Paris). 1996;54(6):235-41. PMID 8949420
- ↑ a , b , c , d , e , f , g , h , i , j et k (en) Samuel M Moskowitz, James F Chmiel, Darci L Sternen, Edith Cheng, and Garry R Cutting, « CFTR-Related Disorders » sur http://www.ncbi.nlm.nih.gov, Created March 26, 2001. Last updated February 19, 2008., GeneReviews. Consulté le 30 mai 2008
- ↑ (en) Sheppard D. Welsh M. Structure and Function of the CFTR Chloride Channel Physiological Reviews Jan 1999 Vol. 79 ; 1 : S23-S45
- ↑ (en) Gadsby D. Nairn A. Control of CFTR Channel Gating by Phosphorylation and Nucleotide Hydrolysis Physiological Reviews Jan 1999 Vol. 79S; 1
- ↑ Physiologie humaine de Lauralee Sherwood et Alain Lockhart édité en 2006 par de boeck Lire l'ouvrage
- ↑ a , b et c (fr) Jean-Jacques Robert, « Diabète de la mucoviscidose », dans Médecine thérapeutique / Pédiatrie, vol. 8, no 3, mai-juin 2005, p. 217-24 [texte intégral (page consultée le 19 juin 2008)]
- ↑ a , b , c , d , e , f , g , h , i , j , k , l , m , n , o et p (fr) Prise en charge du patient atteint de mucoviscidose - Pneumologie et infectiologie, Recommandations de la Société française de pédiatrie (SFP), 2002.
- ↑ a et b (en) Saiman L. Microbiology of early CF lung disease. Paediatr Respir Rev. 2004;5 Suppl A:S367-9. PMID 14980298
- ↑ (en) Girish D Sharma, « Cystic Fibrosis » sur http://www.emedicine.com, 2006. Consulté le 18 juin 2008
- ↑ a , b , c , d , e , f , g , h et i (fr) M. Roussey, « La mucoviscidose (cours en ligne) » sur http://www.med.univ-rennes1.fr, 18 février 2000. Consulté le 19 juin 2008
- ↑ (en) Eggermont E, De Boeck K. Small-intestinal abnormalities in cystic fibrosis patients. Eur J Pediatr. 1991 Oct;150(12):824-8. Review. PMID 1743211
- ↑ (en) Kulczycki LL, Shwachman H. Studies in cystic fibrosis of the pancreas; occurrence of rectal prolapse. N Engl J Med. 1958 Aug 28;259(9):409-12. PMID 13578072
- ↑ (en) Holsclaw DS, Perlmutter AD, Jockin H, Shwachman H (1971) Genital abnormalities in male patients with cystic fibrosis. J Urol. 1971 Oct;106(4):568-74. PMID 4399160
- ↑ (en) Maldonado M, Martinez A, Alobid I, Mullol J. The antrochoanal polyp. Rhinology. 2004 Dec;42(4):178-82. Review. PMID 15626248
- ↑ (en) Ramsey B, Richardson MA. Impact of sinusitis in cystic fibrosis. Allergy Clin Immunol. 1992 Sep;90(3 Pt 2):547-52. PMID 1527348
- ↑ (en) Ballestero Y, Hernandez MI, Rojo P, Manzanares J, Nebreda V, Carbajosa H, Infante E, Baro M. Hyponatremic dehydration as a presentation of cystic fibrosis. Pediatr Emerg Care. 2006 Nov;22(11):725-7. PMID 17110865
- ↑ (en) Wiebicke W, Artlich A, Gerling I. Myocardial fibrosis--a rare complication in patients with cystic fibrosis. Eur J Pediatr. 1993 Aug;152(8):694-6. PMID 8404977
- ↑ a et b (en) Rosenstein BJ, Cutting GR for the Cystic Fibrosis Consensus Panel. The diagnosis of cystic fibrosis: A consensus statement. J Pediatr 1998; 132: 589-595. PMID: 9580754
- ↑ (en) Mishra A, Greaves R, Massie J, « The Relevance of Sweat Testing for the Diagnosis of Cystic Fibrosis in the Genomic Era », dans Clin Biochem Rev., vol. 26, 2005 Nov, p. 135-53 [résumé, texte intégral (pages consultées le 26 juin 2008)]
- ↑ a , b , c et d M. Marchand, C. Jarreau, M. Chauffert, I. Garcia, D. Asselin, J.-P. Thouvenot, A.-F. Genest., « The sweat test », dans Annales de Biologie Clinique. Volume 56, Number 2, 215-21, Mars - Avril 1998, Pratique quotidienne, vol. 56, no 2, Mars - Avril 1998, p. 215-21 [texte intégral (page consultée le 25 mai 2008)]
- ↑ (en) Green A, Dodds P, Pennock C. A study of sweat sodium and chloride, criteria for the diagnosis of cystic fibrosis. Ann Clin Biochem 1985;22:171-6. PMID: 4004107
- ↑ (fr) Nguyen Hkhoa, « test de la sueur » sur bioch.ap-hop-paris.fr
- ↑ (fr) Marc Bellaïche, Jerome Viala, A.Blanc, C.Beyler, A.Mollet, Internat médecine Pediatrie, Vernazobres Grego, collection intermed, 1997, 175-194 p. (ISBN 2-84136-065-2)
- ↑ (en) Augarten A, Hacham S, Kerem E, Kersm BS, Szeinberg A, Laufer J, Doolman R, Altshuler R, Blau H, Bentur L, Gazit E, Katznelson D, Yahar Y. The significance of sweat Cl/Na ratio in patients with borderline sweat test. Pediatr.Pulmonol, 1995; 20: 369-371. PMID: 8649916
- ↑ (en) Desmarquest P, Feldmann D, Tamalat A et al. Genetype analysis and phenotypic manifestations of children with intermediate sweat chloride test results. Chest 2000; 118: 1591- 1597. PMID: 11115444, Résumé, Texte complet
- ↑ (en) Gilfillan A, Warner JP, Kirk J et al. P67L: a cystic fibrosis allele with mild effects found at high frequency in the Scottish population. J.Med.Gent. 1998; 35: 122-125. Résumé
- ↑ (en) Highsmith WE, Burch LH, Zhou Z, et al., « A novel mutation in the CF gene in patients with pulmonary diseases but normal sweat chloride concentration. », dans N Engl J Med, vol. 331, no 15, 1994, p. 974-80 [résumé]
- ↑ (en) Augarten A, Kerem B-S, Yahav Y, Noiman S, Rivlin Y, Tal A, Blau H, Ben-Tur L, Szeinberg A, Kerem E, Gazit E. Mild cystic fibrosis and normal or borderline sweat test in patients with the 3849+10kb C®T mutation. Lancet 1993; 342; 25-26. PMID: 8100293
- ↑ (en) Strong TV, Smit LS, Turpin SV, Cole JL, Tom Hon C, Markiewicz D, Petty TL, Craig MW, Rosenow EC, Tsui L-C, Lannuzzi MC, Knowles MR and Collins FS. Cystic fibrosis gene mutation in two sisters with mild disease and normal sweat electrolyte levels. N Engl J Med, 1991; 325; 1630-34. Lien
- ↑ (en) Stewart B, Zabner J, Shuber AP, Welsh MJ, McCray PB. Normal sweat chloride values do not exclude the diagnosis of cystic fibrosis. Am J Respir Crit Care Med 1995; 151: 899-903. Résumé
- ↑ (en) Cystic Fibrosis Foundation. Patient Registry 1994. Annual Data Report. Bethesda (MD). The Foudation, August 1995.
- ↑ (en) Knowles MR, Paradiso AM, Boucher RC (1995) In vivo nasal potential différence: techniques and protocols for assessing efficacy of gene transfer in cystic fibrosis. Hum Gene Ther 6:445-55 PMID 7542031
- ↑ (en) Schuler D, Sermet-Gaudelus I, Wilschanski M, Ballmann M, Dechaux M, Edelman A, Hug M, Leal T, Lebacq J, Lebecque P, Lenoir G, Stanke F, Wallemacq P, Tummler B, Knowles MR. Basic protocol for transepithelial nasal potential difference measurements. J Cyst Fibros 3 Suppl. 2004; 2: 151–5. PMID: 15463949
- ↑ (en) Standaert TA, Boitano L, Emerson J, Milgram LJ, Konstan MW, Hunter J, Berclaz PY, Brass L, Zeitlin PL, Hammond K, Davies Z, Foy C, Noone PG, Knowles MR. Standardized procedure for measurement of nasal potential difference: an outcome measure in multicenter cystic fibrosis clinical trials. Pediatr Pulmonol. 2004; 37: 385–92. PMID: 15095320
- ↑ a et b (fr) Labbé A, Bellon G, Héraud MC, Arbre P, Goddon G, Alton E : Détermination des différences de potentiel transépithélial (DDPTE) nasal dans la mucoviscidose. Arch Fr Pediatr 1991 ; 48 : 617-20. INIST.
- ↑ (en) Hoffmann T, Böhmer O, Hüls G, Terbrack HG, Bittner P, Klingmüller V, Heerd E, Lindemann H : Conventionnal and modified nasal potential-difference. Measurement in cystic fibrosis. Am J Respir Crit Care Med 1997 ; 155 : 1908-13. PMID: 9196094, Abstract Am.J.Respir.Crit.Care Med.
- ↑ (fr) R. Matran, T. Perez, V. Neve, S. Robin, R. Neviere. Actualités sur les explorations diagnostiques de la mucoviscidose. Revue des maladies respiratoires. ISSN 0761-8425. Congrès La mucoviscidose de l'enfant à l'adulte. Congrès No4, Lille , FRANCE (27/03/2003) 2003, vol. 20, no2, CAHIER1, pp. S21-S25. En ligne
- ↑ (en) Weiss LN. The diagnosis of wheezing in children. Am Fam Physician. 2008 Apr 15;77(8):1109-14. PMID: 18481558
- ↑ (en) F. Charles Hiller. Cystic Fibrosis Masquerading as Asthma and Bronchitis in Young Adults. Pediatric Asthma, Allergy & Immunology. December 1, 2003, 16(4): 319-324. doi:10.1089/088318703322751363. En ligne
- ↑ (en) Burge D, Drewett M. Meconium plug obstruction. Pediatr Surg Int. 2004 Feb;20(2):108-10. Epub 2004 Feb 4.Click here to read 14760494
- ↑ (en) Keckler SJ, St Peter SD, Spilde TL, Tsao K, Ostlie DJ, Holcomb GW 3rd, Snyder CL. Current significance of meconium plug syndrome. J Pediatr Surg. 2008 May;43(5):896-8. PMID: 18485962
- ↑ (en) Lozada-Muñoz L, Aliaga MD. Shwachman-Diamond syndrome: the clinical imitator of cystic fibrosis. P R Health Sci J. 1995 Dec;14(4):275-7. PMID: 8637967
- ↑ (en) Conklin LS, Zeitlin PL, Cuffari C. Cystic Fibrosis presenting as recurrent pancreatitis in a young child with a normal sweat test and pancreas divisum: a case report. J Med Case Reports. 2008 May 23;2(1):176. PMID: 18501000
- ↑ (en) Petrone MC, Arcidiacono PG, Testoni PA. Endoscopic ultrasonography for evaluating patients with recurrent pancreatitis. World J Gastroenterol. 2008 Feb 21;14(7):1016-22. PMID: 18286681
- ↑ (en) Dalcin Pde T, Abreu E Silva FA. Cystic fibrosis in adults: diagnostic and therapeutic aspects. J Bras Pneumol. 2008 Feb;34(2):107-17. PMID: 18345455
- ↑ a et b (fr) Grosskofpf C, Farriaux JP, Vidailhet M, Briard ML, Navarro J, Turck D, Travert G, Belot V, Bloch J, Roussel P. «Le programme national de dépistage néonatal de la mucoviscidose: mise en place et organisation». Archives de Pédiatrie 2003; 10(Suppl 2): S364-369. Inist
- ↑ a , b et c (fr) E Deneuville et M Roussey. Le dépistage néonatal de la mucoviscidose en France et dans le monde.Organisation, bénéfices, difficultés. État des lieux en 2006. Journées de Techniques Avancées en Gynécologie et Obstétrique, JTA 2007. En ligne, Résumé de l'État des lieux en 2007
- ↑ Décret no 2000-570 du 23 juin 2000 fixant les conditions de prescription et de réalisation des examens des caractéristiques génétiques d'une personne et de son identification par empreintes génétiques à des fins médicales et modifiant le code de la santé publique. En ligne sur legifrance.gouv.fr
- ↑ a , b , c , d et e (fr) E.Flori et B.Doray, « Mucoviscidose » sur http://www-ulpmed.u-strasbg.fr, Février 2007, Faculté de médecine de Strasbourg - Item 31 - 2006/2007. Consulté le 18 juin 2008
- ↑ (en) Casaccia G, Trucchi A, Nahom A, Aite L, Lucidi V, Giorlandino C, Bagolan P., « The impact of cystic fibrosis on neonatal intestinal obstruction: the need for prenatal/neonatal screening », dans Pediatr Surg Int, vol. 1-2, no 19, 2003 Apr, p. 75-8 [résumé (page consultée le 19 juin 2008)]
- ↑ a et b (fr) Avis n° 83 du comité consultatif national d'éthique sur le dépistage prénatal généralisé de la mucoviscidose datant du 25 mars 2004.
- ↑ (fr) J Lansac, F Guérif AMP l'assistance médicale à la procréation en pratique voir l'ouvrage p315
- ↑ (en) Four decades at a glance sur http://www.cysticfibrosis.ca, 5 aout 2004, Canadian Cystic Fibrosis Foundation. Consulté le 30 mai 2008
- ↑ Définition : la vie médiane ou encore vie probable, est une donnée statistique exprimant pour un enfant né à une date donnée, la durée de vie qu'il a une chance sur deux de dépasser, si les conditions de mortalité demeurent inchangées.
- ↑ (fr) Fondation canadienne de la fibrose kystique, Rapport du Registre canadien de données sur les patients, Toronto, Ontario, 2002
- ↑ (en) Andrew Fogarty, Richard Hubbard, John Britton. International Comparison of Median Age at Death From Cystic Fibrosis. Chest. 2000;117:1656-1660. « The international median age at death increased from 8 years in 1974 to 21 years in 1994 ».
- ↑ (en)New Statistics Show CF Patients Living Longer, sur http://www.cff.org/ le site de la Cystic Fibrosis Foundation, 2006 news, posted 04/26/06. « The median age of survival for cystic fibrosis (CF) patients has risen to 36.8 years, up from the previous year’s figure of 35.1 years, according to new data released by the Cystic Fibrosis Foundation. »
- ↑ a , b , c , d , e , f , g et h (fr) Protocole national de diagnostic et de soins : Mucoviscidose. sur http://www.has-sante.fr, novembre 2006, Haute Autorité de Santé. Consulté le 18 juin 2008
- ↑ a , b , c , d , e , f , g , h , i , j , k et l (fr) Prise en charge du patient atteint de mucoviscidose - Observance, nutrition, gastro-entérologie et métabolisme, Recommandations de la Société Française de Pédiatrie
- ↑ (en) Drenkard, E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microb. Infect. 2003. 5:1213-1219. PMID: 14623017
- ↑ (en) Jehanne M. Treatment of superinfections caused by pyocyanic bacillus in patients with mucoviscidosis. Efficacy of cefsulodin in combination with an aminoglycoside Pathol Biol (Paris). 1989 May;37(5):500-3. PMID 2780107
- ↑ (en) Lenoir G, Antypkin YG, Miano A, Moretti P, Zanda M, Varoli G, Monici Preti PA, Aryayev NL. Efficacy, safety, and local pharmacokinetics of highly concentrated nebulized tobramycin in patients with cystic fibrosis colonized with Pseudomonas aeruginosa. Paediatr Drugs. 2007;9 Suppl 1:11-20. PMID 17536871
- ↑ (en) Jafri HS, McCracken GH Jr. Fluoroquinolones in paediatrics. Drugs. 1999;58 Suppl 2:43-8. PMID 10553704
- ↑ (en) Mirakhur A, Gallagher MJ, Ledson MJ, Hart CA, Walshaw MJ.Fosfomycin therapy for multiresistant Pseudomonas aeruginosa in cystic fibrosis. J Cyst Fibros. 2003 Mar;2(1):19-24. PMID 15463841
- ↑ (en) Szaff M, Hoiby N. Antibiotic treatment of Staphylococcus aureus infection in cystic fibrosis. Acta Paediatr Scand. 1982 Sep;71(5):821-6.PMID 7180452
- ↑ (fr) La Dornase alfa, avis de la Haute Autorité de Santé française sur has santé.fr
- ↑ a , b et c (fr) Christiane Knoop (Hôpital Erasme - Institut de Mucoviscidose de l'ULB), « Transplantation pulmonaire pour mucoviscidose », dans Kinéréa, Société de kinésithérapie de réanimation, Paris, France, no 37, 2003, p. 58-63 (ISSN 0999-9183) [résumé, texte intégral (pages consultées le 13 juin 2008)]
- ↑ (en) James R. Yankaskas MD, George B. Mallory Jr. MD and Consensus Committee, « Lung Transplantation in Cystic Fibrosis : Consensus Conference Statement », dans Chest, no 113, 1998, p. 217-226 [résumé, texte intégral (pages consultées le 16 juin 2008)]
- ↑ (en) Yung MW, Gould J, Upton GJ. Nasal polyposis in children with cystic fibrosis: a long-term follow-up study. Ann Otol Rhinol Laryngol. 2002 Dec;111(12 Pt 1):1081-6. PMID 12498368
- ↑ (en) Gysin C, Alothman GA, Papsin BC. Sinonasal disease in cystic fibrosis: clinical characteristics, diagnosis, and management. Pediatr Pulmonol. 2000 Dec;30(6):481-9. PMID 11109061
- ↑ (en) Moss RB, King VV. Management of sinusitis in cystic fibrosis by endoscopic surgery and serial antimicrobial lavage. Reduction in recurrence requiring surgery. Arch Otolaryngol Head Neck Surg. 1995 May;121(5):566-72. PMID 7727092
- ↑ (en) Kelvin H.-V. Tan, Michael Mulheran, Alan J. Knox and Alan R. Smyth, « Aminoglycoside Prescribing and Surveillance in Cystic Fibrosis », dans American Journal of Respiratory and Critical Care Medicine, vol. 167, 2003, p. 819-823 [résumé, texte intégral (pages consultées le 16 juin 2008)]
- ↑ (en) Sawka MN, Montain SJ. Fluid and electrolyte supplementation for exercise heat stress. Am J Clin Nutr. 2000 Aug;72(2 Suppl):564S-72S. PMID 10919961 ajcn.org
- ↑ (en) Kriemler S, Wilk B, Schurer W, Wilson WM, Bar-Or O. Preventing dehydration in children with cystic fibrosis who exercise in the heat. Med Sci Sports Exerc. 1999 Jun;31(6):774-9. PMID 10378902
- ↑ (en) Desmazes-Dufeu N, Hubert D, Burgel PR, Kanaan R, Velea V, Dusser D. Severe dehydration and August 2003 heat wave in a cohort of adults with cystic fibrosis Presse Med. 2005 May 14;34(9):647-8. PMID 15988338
- ↑ (en) Nixon PA, Orenstein DM, Kelsey SF, Doershuk CF. The prognostic value of exercise testing in patients with cystic fibrosis. N Engl J Med 1992 ; 327 : 1785-8. Résumé
- ↑ a et b (en) Schneiderman-Walker J, Pollock SL, Corey M, Wilkes DD, Canny GJ, Pedder L, Reisman JJ : A randomized controlled trial of a 3year home exercise program in cystic fibrosis. J Pediatr 2000 ; 136 : 304-10. PMID: 10700685
- ↑ a et b (en) de Jong W, Grevink RG, Roorda RJ, Kaptein AA, van der Schans CP : Effect of a home exercise training program in patients with cystic fibrosis. Chest 1994 ; 105 : 463-8. PMID: 8306748
- ↑ (en) Moorcroft AJ, Dodd ME, Morris J, Webb AK. Individualised unsupervised exercise training in adults with cystic fibrosis: a 1 year randomised controlled trial. Thorax. 2004 Dec;59(12):1074-80. PMID: 15563708
- ↑ (en) Selvadurai HC, Blimkie CJ, Meyers N, Mellis CM, Cooper PJ, Van Asperen PP. Randomized controlled study of in-hospital exercise training programs in children with cystic fibrosis. Pediatr Pulmonol. 2002 Mar;33(3):194-200. PMID: 11836799
- ↑ (en) Bradley J, Moran F. Physical training for cystic fibrosis. Cochrane Database Syst Rev 2002 ; (CD002768). PMID: 18254007
- ↑ (en) Stead RJ, Grimmet SFM, Rogers SM, Back DJ, Orme ML, Hodson ME, Batten JC Pharmacokinetics of contraceptive steroids in patients with cystic fibrosis. Thorax 1987 Jan;42(1):59-64. PMID 3112991
- ↑ (en) Siegel B, Siegel S. Pregnancy and delivery in a patient with cystic fibrosis of the pancreas. Obstet Gynecol 1960; 16:438-440
- ↑ (en) Kent, NE, Farquaharson, DF, « Cystic fibrosis in pregnancy », dans Canadian Medical Association Journal, vol. 149, no 6, 1993, p. 809-813 [résumé (page consultée le 29 mai 2008)]
- ↑ (en) Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE., « Pregnancy in Cystic Fibrosis : Fetal and Maternal Outcome », dans Chest, vol. 118, no 1, 2000, p. 85-91 [résumé, texte intégral (pages consultées le 29 mai 2008)]
- ↑ (en) Goss, CH, Rubenfeld, GD, Otto, K, et al, « The Effect of Pregnancy on Survival in Women With Cystic Fibrosis », dans Chest, vol. 124, 2003, p. 1460-1468 [résumé, texte intégral (pages consultées le 29 mai 2008)]
- ↑ (en) Scott JR, Wagoner L, Olsen S, Taylor DO, Renlund DG. Pregnancy in heart transplant recipients: management and outcome. Obstet Gynecol. 1993 Sep;82(3):324-7. PMID 8355928
- ↑ (fr)[pdf] Texte de la circulaire relative à l’organisation des soins pour la prise en charge des patients atteints de mucoviscidose.
- ↑ (fr) Albert-Gérard Logeais. Mucoviscidose : quelle est la place des associations de malades dans la nouvelle organisation des soins ? La Revue du Praticien 2003,53:121-122. INIST.
- ↑ (en) About the Cystic Fibrosis Foundation sur http://www.swamppopmusicfest.com.
- ↑ (en) www.cff.org/ site de la Cystic Fibrosis Foundation
- ↑ Le site de l'European Cystic Fibrosis Society en donne une liste non exhaustive sur ses pages web voir (en) www.ecfsoc.org.
- ↑ (fr) [pdf] : Prise en charge de la mucoviscidose en affection de longue durée, point de repère, mars 2007, sur http://www.ameli.fr
- ↑ (en) Lieu TA, Ray GT, Farmer G, Shay GF. The cost of medical care for patients with cystic fibrosis in a health maintenance organization. Pediatrics. 1999 Jun;103(6):e72 PMID 10353969
- ↑ (en) Johnson JA, Connolly MA, Jacobs P, Montgomery M, Brown NE, Zuberbuhler P. Cost of care for individuals with cystic fibrosis: a regression approach to determining the impact of recombinant human DNase.Pharmacotherapy. 1999 Oct;19(10):1159-66. PMID 10512065
- ↑ Site de l' European Cystic Fibrosis Society sur www.ecfsoc.org
- ↑ Site de la revue Journal of Cystic fibrosis
- ↑ Recommandations de l' European Cystic Fibrosis Society
- ↑ a , b et c (en) Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev 1999 ; 79 : s193-s214. PMID: 9922382, Texte complet
- ↑ (en) Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. : An animal model for cystic fibrosis made by gene targeting. Science 1992 ; 257 : 1083-8. PMID: 1380723, Abstract
- ↑ (en) Stotland PK, Radzioch D, Stevenson MM. Mouse models of chronic lung infection with Pseudomonas aeruginosa: models for the study of cystic fibrosis. Pediatr Pulmonol 2000 ; 30 : 413-24. PMID: 11064433
- ↑ a et b (fr) Un modèle murin mimant les manifestations pulmonaires de la mucoviscidose vient d’être mis au point. Revue des Maladies Respiratoires. Vol 22, n° spécial - juin 2005. pp. 11-19. Doi : RMR-06-2005-22-6-0761-8425-101019-200505321. Texte intégral
- ↑ (en) M Mall , BR G Rubb , JR Harkeam , WK O’Neal , RC Boucher. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 2004 ; 10 : 487-93. PMID: 15077107, Abstract
- ↑ Kennedy MJ. Current status of gene therapy for cystic fibrosis pulmonary disease Am J Respir Med. 2002;1(5):349-60. PMID 14720037
- ↑ (en) Weiss DJ, Pilewski JM, The Status of gene therapy for cystic fibrosis Semin Respir Crit Care Med. 2003 Dec;24(6):749-70. PMID 16088590
- ↑ (en) Cheng SH, Fang SL, Zabner J, Marshall J, Piraino S, Schiavi SC, Jefferson DM, Welsh MJ, Smith AE. Functional activation of the cystic fibrosis trafficking mutant delta F508-CFTR by overexpression. Am J Physiol. 1995 Apr;268(4 Pt 1):L615-24. PMID 7733303
- ↑ (en) Zhang XM, Wang XT, Yue H, Leung SW, Thibodeau PH, Thomas PJ, Guggino SE. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2003 Dec 19;278(51):51232-42. Epub 2003 Oct 7. PMID 14532265
- ↑ (fr) RCP Zavesca(R) Miglustat sur http://www.agmed.sante.gouv.fr
- ↑ (en) Norez C, Noel S, Wilke M, Bijvelds M, Jorna H, Melin P, DeJonge H, Becq F. Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the alpha-glucosidase inhibitor miglustat. FEBS Lett. 2006 Apr 3;580(8):2081-6. Epub 2006 Mar 10. PMID 16546175
- ↑ Lisa Bentley Triumphs Over CF, 7 juillet 2004, Canadian Cystic Fibrosis Foundation. Consulté le 26 janvier 2007
- ↑ CricInfo: Chris Davies, août 2001, CricInfo.com. Consulté le 26 janvier 2007
- ↑ Stephen Holden, « An artist whose medium was pain », 3 avril 1997, New York Times. Consulté le 23 février 2007
- ↑ AC Player Battles Disease To Play Basketball, 28 janvier 2005, Anderson University. Consulté le 18 juillet 2007
- ↑ Just-Gregory.net: Fanzone: Biographie, Just-Gregory.com. Consulté le 26 janvier 2007
- ↑ Becky Barrow, « Pop singer dies after valiant battle with cystic fibrosis », 3 juillet 2003, Telegraph. Consulté le 23 février 2007
- ↑ Online World of Wrestling: Wrestler Profile: Andrew Simmons, onlineworldofwrestling.com. Consulté le 1er février 2007
- ↑ Naked Before God: The Return of a Broken Disciple, Anglican Theological Review (été 1999). Consulté le 23 février 2007.
- ↑ Chopin et la mucoviscidose sur Liberation.fr
Voir aussi
Articles connexes
Liens externes
 Portail de la médecine
Portail de la médecine Portail des soins infirmiers et de la profession infirmière
Portail des soins infirmiers et de la profession infirmière

La version du 23 juillet 2008 de cet article a été reconnue comme « article de qualité », c'est-à-dire qu'elle répond à des critères de qualité concernant le style, la clarté, la pertinence, la citation des sources et l'illustration. Catégories : Article de qualité | Maladie génétique | Maladie génétique congénitale | Canalopathie | Pédiatrie

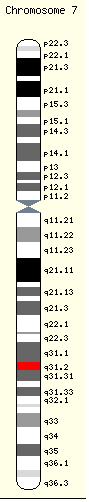

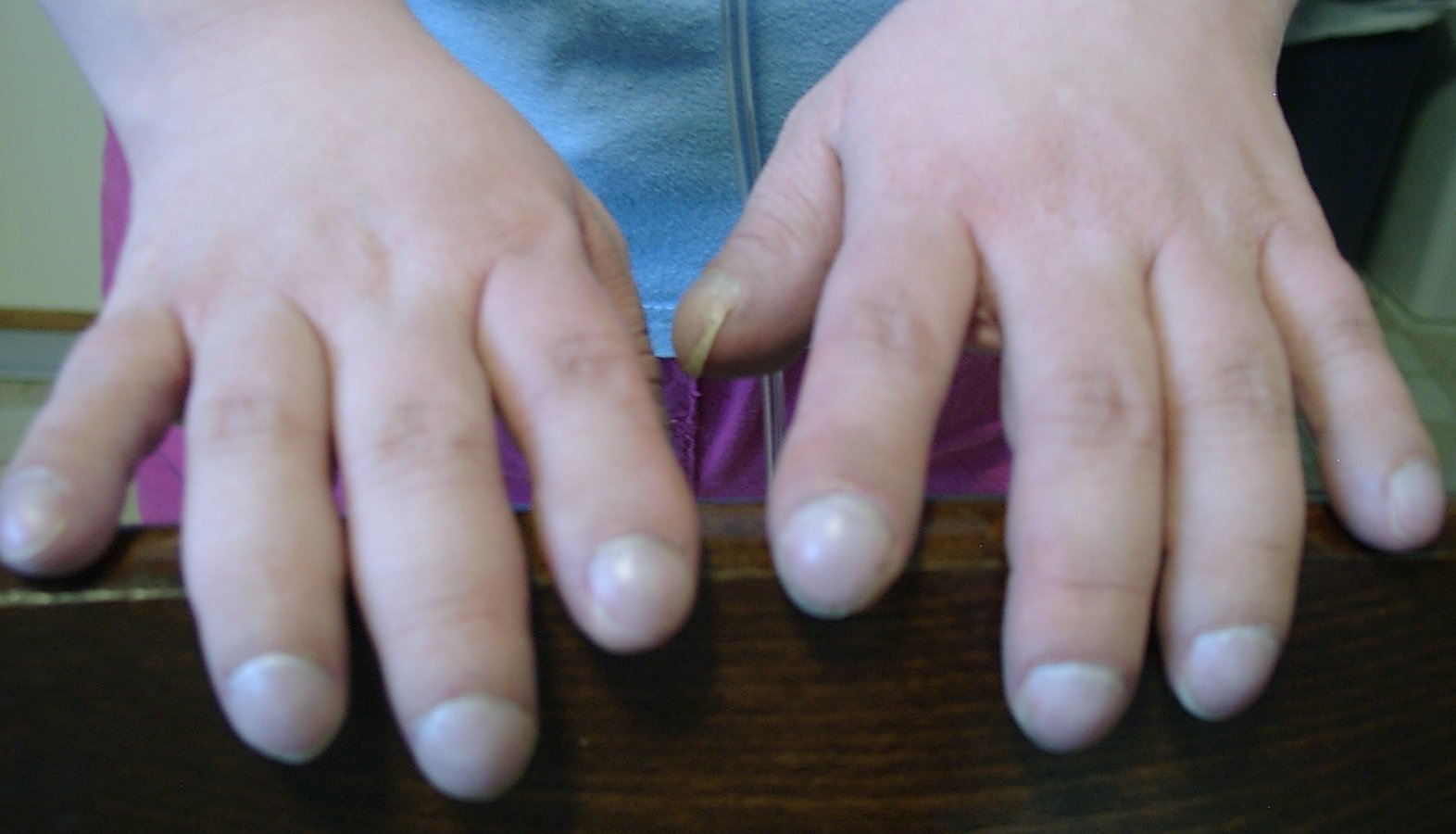



Wikimedia Foundation. 2010.