- Maladie de Hirschsprung
-
Maladie de Hirschsprung
Classification et ressources externes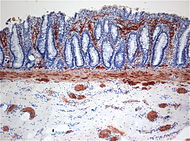
Histopathologie / Maladie de Hirschsprung. Présence aberrante de fibres positives à l'enzyme Acétylcholine-estérase ou ACHE) (en brun) dans les lamina propria mucosae CIM-10 Q43.1 CIM-9 751.3 OMIM 142623 DiseasesDB 5901 MedlinePlus 001140 eMedicine med/1016 MeSH D006627 La maladie de Hirschsprung est une anomalie de fonctionnement de la partie terminale de l’intestin se traduisant par une constipation ou une occlusion intestinale. Cette anomalie est le résultat de l’absence de développement congénital des cellules neuroganglionnaires assurant la transmission des informations nécessaires à la régulation intestinale.
Cette maladie est considérée comme une neurocristopathie ou maladie dérivant des crêtes neurales.
Cette maladie peut être isolée ou s'inscrire dans un syndrome.
Dans les formes non syndromiques, plusieurs gènes ont été trouvés responsables de cette maladie.
Sommaire
Autres noms de la maladie
- Aganglionose digestive totale
- Maladie de Hirschsprung type 1 forme dominante
- Maladie de Hirschsprung type 2 forme récessive
- Maladie de Hirschsprung type 3
- Mégacolon congénital
Incidence
La Maladie de Hirschsprung est une maladie génétique rare qui touche 1 naissance sur 5000, en majorité des garçons (80%). Il s'agit bien souvent de formes sporadiques (sans antécédent familial) mais on connaît des formes familiales.
Description
La maladie de Hirschsprung est une formation congénitale touchant le plexus nerveux intrinsèque qui est anormalement absent de la paroi digestive depuis l’anus jusqu’à une hauteur variable de l’intestin, généralement la partie moyenne du sigmoïde.
Cette maladie est due à l'absence partielle ou totale de ganglions nerveux, dont le rôle est de permettre le bon fonctionnement des muscles de l'intestin (et plus particulièrement du côlon). De l'existence de ce segment aganglionnaire résulte une paralysie intestinale, se traduisant généralement par une occlusion fonctionnelle, parfois simplement par une constipation importante.
La majorité des enfants ont des retards d’émission du méconium, l'émission ne survenant qu’après plus de 48 heures de vie néonatale. Les signes cliniques sont digestifs : constipation, distension abdominale ou vomissement ou entérocolite. Mais 10% des diagnostics de maladie de Hirschprung ne sera fait qu’après l’âge de 1 an (dans les formes non syndromiques).
C’est une maladie polygénique c’est-à-dire due à la mutation de plusieurs gènes indépendants, dont une combinaison particulière détermine cette malformation.
Il est à noter que cette maladie existe aussi chez plusieurs animaux, notamment chez le rat (ce qui facilite la recherche en laboratoire sur cette maladie). Il semblerait que chez cette espèce l'anomalie génétique comprend également une dépigmentation du pelage. Il en est de même chez le cheval.
Formes cliniques
Il existe différentes formes de la Maladie de Hirschsprung, dont la gravité dépend de la longueur du segment aganglionnaire. Dans 90% des cas, il s'agit de la forme "classique" qui touche simplement la terminaison du colon (le rectum et le colon sigmoïde).
Il existe aussi des formes courtes n'intéressant que la partie basse du rectum et à l'opposé des formes longues pouvant atteindre tout le colon voire remonter plus ou moins sur l'intestin grêle.
En fonction de l’étendue de l’atteinte de la maladie au niveau intestinal, on distingue :
- Hirschsprung court segment : localisée au sigmoïde dans 80 % des cas.
- Hirschsprung long segment : étendue au colon dans 15 % des cas.
- Hirschsprung totale : localisée à tout l’intestin dans 5 % des cas.
Diagnostic
Le diagnostic a pu être évoqué sur les échographies ante-natales du fait de la dilatation intestinale. À la naissance il existe une occlusion intestinale avec ballonnement abdominal souvent considérable puis vomissement bilieux (vert). Le méconium (premières selles très noires évacuées normalement durant les premières 24h) est ici retardé et anormalement prolongé. Le passage en douceur d'une grosse sonde rectale permet pratiquement d'affirmer le diagnostic lorsqu'elle ramène des selles et des gaz en abondance en déballonnant l'enfant en même temps. Cela se produit si la sonde a pu être montée jusque dans le colon dilaté en amont de la zone aganglionnaire.
Repose essentiellement sur la biopsie intestinale du rectum qui montre l'hypertrophie des fibres nerveuses parasympathiques et l'absence de cellule ganglionnaire des plexus de Meissner
Diagnostic différentiel
- POIC : pseudo-obstruction intestinale chronique - accidents sub-occlusifs à répétition (neuropathies digestives, myopathie)
- Syndrome du bouchon méconial : bilans Hirschsprung et mucoviscidose négatifs (test de la sueur, biopsies) - amélioration spontanée.
- Microcôlon gauche : terrain : gros bébés, mère diabétique.
Étiologie
Formes non syndromiques
6 gènes ont été identifiés comme responsables de maladie de Hirschsprung :
- RET situé sur le locus q11.2 du chromosome10. Gène le plus souvent impliqué dans les formes familiales de maladie de Hirschsprung.164761 (en) (rearranged during transfection protooncogene)
- GDNF situé sur le locus p13.1-p12 du chromosome 5
- NRTN situé sur le locus p13.3 du chromosome 19
- EDNRB situé sur le locus q22 du chromosome 13
- EDN3 situé sur le locus q13.2-q13.3 du chromosome 20
- ECE1 situé sur le locus p36.1 du chromosome 1
Formes génétiques syndromiques
MALADIES GÉNÉTIQUES COMPORTANT UNE MALADIE DE HIRSCHSPRUNG Syndromes Signes cliniques Transmission Chromosome
locusGène Fréquence Numéro MIM Syndrome de Bardet-Biedl Retard mental
Obésité
Polydactylie
Rétinite pigmentaire
Anomalies rénales et génitalesRécessive 7 loci connus 2-10% 209900 Chondrodysplasie métaphysaire autosomique récessive Nanisme
Cheveux rare
Déficit immunitaireRécessive 9 p13 RMRP 7-9% 250250 Syndrome d'Ondine Hypoventilation
NeuroblastomeDominante PHOX2B 16% 209880 Dysautonomie familiale Récessive 9 q31 IKBKAP ? [1] Syndrome de Mowat-Wilson Microcéphalie
Retard mental
Agénésie du corps calleux
Cardiopathie congénitale
Épilepsie
Petite tailleDominante 2q22 ZFHX1B 62-70% 235730 Pseudo-obstruction intestinale chronique idiopathique Dominante et Récessive ? ? Jusque 20 % 243180 Syndrome de Goldberg-Shprintzen Récessive 10 q21.1 KIAA1279 609460 Néoplasie endocrinienne multiple type 2 Dominante RET Rare 171400 Neurofibromatose type 1 Dominante Inconnu 162200 Syndrome de Smith-Lemli-Opitz Retard mental
Syndactylie
Cardiopathie
Anomalies sexuellesRécessive 11 q12-q13 DHCR7 Inconnu 270400 Syndrome de Waardenburg-Shah Dominante et Récessive Presque 100% [2] Syndrome L1 Retard mental
Agénésie du corps calleux
Hydrocéphalie
Pouce en adductionRécessive à l'X X L1CAM Inconnu [3] - SOX10 dans le syndrome Yemenite 4 situé sur le locus q13.1 du chromosome22 [4]
Chromosomique
- Trisomie 21
- Délétion 10q
- Délétion 13q
- Délétion 2q22
Cause inconnue
Compte pour 10 à 15 % des cas de maladie de Hirschsprung.
Traitement
Il consiste à introduire une sonde dans le rectum ou à effectuer de petits lavements prudents. Ceci a pour but de permettre l'évacuation des selles, mais cette technique n'est pas efficace dans tous les cas. Dans les formes où ce traitement médical est inefficace, on effectue ce que l'on appelle une colotomie [et non pas une colostomie qui est la création d'un anus artificiel par abouchement à la peau d'une portion de côlon]
La colotomie consiste à effectuer une ouverture chirurgicale de la paroi du côlon permettant ainsi l'exploration de celui-ci. Il autorise parfois la découverte d'anomalie mais également l'ablation de petites tumeurs bénignes comme des polypes qui font saillie à l'intérieur de la cavité intestinale. Au cours de l'occlusion intestinale, la colotomie permet la décompression et l'évacuation du côlon. Néanmoins, cette technique présente quelques dangers, en effet, en raison du risque élevé de dissémination des microbes qu'elle entraîne et elle nécessite des précautions particulières pour sa réalisation, qui doit se faire dans de très bonnes conditions d'asepsie
La colotomie est recommandée jusqu'à la correction définitive. Quand le colon est atteint dans son entier, une colostomie est alors nécessaire. L'anastomose iléo-colique est une opération chirurgicale qui consiste à relier la partie terminale de l'intestin grêle au colon. Cette opération permet de rétablir la continuité du tube digestif après avoir effectué une ablation partielle (dans cet exemple, une partie du côlon). Après cette ablation de la portion pathologique du côlon, le segment de l'iléon est relié au segment du côlon qui reste, avec du fil ou des agrafes. L'anastomose iléo-colique n'entraîne généralement pas de conséquences sur le fonctionnement du tube digestif
L'opération est réalisable à partir de six mois, si l'état général de l'enfant le permet. Certains chirurgiens estiment qu'une intervention de ce type peut s'effectuer plus précocement. Quand l'ensemble du côlon est atteint, c'est l'iléon normalement innervé qui doit être amené au niveau du rectum ou même de l'anus. Autrement dit, le but recherché est de supprimer les zones intestinales ne contenant plus de plexus de Meissner et d'Auerbach, et de relier les intestins qui fonctionnent normalement à la partie terminale du tube digestif, c'est-à-dire le rectum, si celui-ci possède des plexus " en état de marche ", sinon à l'anus.
Il est important de faire garder ensuite à l'enfant un régime alimentaire à forte teneur en fibres et liquides, afin d'éviter la constipation.
Dans certain cas, des signes opposés comme de l'incontinence ou des souillures peuvent apparaître après une opération. Ils s'améliorent avec une rééducation périnéale à partir des 5 ou 6 ans de l'enfant.
Sources
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 142623
- (en) Melissa A Parisi, Hirschsprung Disease Overview dans GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2005.
- (fr) North American Society for Pediatric Gastroenterology, Hepatology and Nutrition : La maladie de Hirschsprung.
Voir aussi
Articles connexes
Liens externes
Catégories :- Maladie génétique de l'appareil digestif
- Maladie du côlon et du rectum

Wikimedia Foundation. 2010.