- Hémochromatose génétique
-
Hémochromatose génétique
Classification et ressources externes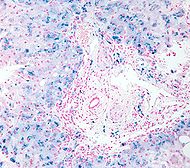
Micrographie d'une hémosidérose. Biopsie hépatique. CIM-10 R79.0 CIM-9 790.6 DiseasesDB 5581 MeSH D019190 Hémochromatose génétique Référence MIM 235200 Transmission Autosomique Récessive Chromosome 6p21.3 Gène HFE Mutation Ponctuelle Mutation de novo Non Nombre d'allèles pathologiques 2 connus Porteur sain 1/10 Prévalence 1 sur 200 à 400 Pénétrance Basse Diagnostic prénatal Possible Liste des maladies génétiques à gène identifié modifier 
L’hémochromatose génétique de type 1 ou hémochromatose classique, par mutation du gène HFE, est une maladie génétique caractérisée par une hyperabsorption du fer par l'intestin entraînant son accumulation dans l'organisme, préférentiellement au niveau de certains tissus et organes. C'est l'une des maladies génétiques les plus fréquentes dans les populations originaires d'Europe du Nord.
Il existe d'autres formes d'hémochromatose primitives ou génétiques (types 2a, 2b, 3 et 4), en particulier des formes pouvant s'exprimer dès l'enfance (hémochromatoses dites juvéniles) mais il s'agit de maladies rarissimes. L'hémochromatose dite secondaire est le plus souvent due à des transfusions répétées de globules rouges.
Sommaire
Historique et Cause
L'hémochromatose génétique de type I est due à une mutation du gène HFE situé sur le locus p21.3 du chromosome 6. Ce gène HFE C282Y a donc été découvert aux États-Unis en 1996 par l' équipe du professeur J.N. Feder[1].
Cette mutation principale, C282Y responsable de la plupart des cas d’hémochromatose génétique en Europe (HFE C282Y) semble avoir été, à l'origine, comme un événement unique, et serait apparue il-y- a environ 4000/4500 ans soit vers -2000 ans avant J.C. sur le chromosome transportant HLA-A3 et B7 d'un habitant Celte de l'Europe préhistorique.
Elle est donc souvent décrite comme une mutation celtique dont on pense que l’origine se situe dans la Préhistoire, à l' époque du Néolithique, dans une population celte située autrefois en Europe centrale et dont la diffusion vers l'ouest et le nord de l’Europe s’est produite par les mouvements de population.
Il a également été suggéré que les migrations des Vikings ont été, bien plus tard, durant le haut Moyen Âge, au cours de "la période Viking " 793-1066, en grande partie responsable de la distribution de cette mutation néolithique durant leurs expéditions lointaines dans des contrées aussi diverses que les pays riverains de la Mer Méditerranée, la Russie ou les pays traversés par les grands fleuves qu' ils ont parcouru, comme le Danube.
Les premières estimations de l'âge de la mutation sont compatibles avec une ou l'autre de ces suggestions. Si on examine les éléments de preuves au sujet de la mutation HFE C282Y fréquence élargie impliquant les haplotypes HLA-A et-B allèles, la validité des calculs sur l’ époque de la mutation, l’ avantage sélectif et les vues actuelles sur l'importance des migrations de population et l'adoption-diffusion (changement culturel) durant la période de transition du néolithique en Europe et depuis lors. nous concluons que la mutation C282Y HFE a eu lieu sur le continent européen vers -2000/-2500ans avant Jésus-Christ.La période néolithique en Europe a marqué le passage de l'économie de chasseurs-cueilleurs du Mésolithique dont l’ alimentation était riche en viande rouge à celle des agriculteurs du néolithique dont l'alimentation constituée principalement de semences de céréales avait un fer réduit. Ce changement de régime alimentaire a probablement provoqué une augmentation de l'incidence de l'anémie ferriprive, en particulier chez les femmes en âge de procréer. L’ hypothèse la plus probable est que l'hémochromatose héréditaire et en particulier celle du cadre HFE mutation du gène C282Y représentent une adaptation à la diminution du fer générée par l’ alimentation à base de céréales du néolithique. Les deux homozygotes et hétérozygotes porteurs de la mutation C282Y HFE ont augmenté les réserves de fer et possédait donc une adaptation avantageuse dans les conditions de vie agraires du néolithique. Une estimation de l’ époque et du lieu d’origine de la mutation C282Y HFE qui a conclu à la situer au début du Néolithique en Europe du Nord est donc compatible avec cette hypothèse. Il est donc vraisemblable que cette mutation C282Y ait été sélectionnée au cours de l'évolution car sa présence réduisait la fréquence de la carence en fer, fréquente à ces époques, et augmentait ainsi la fécondité des femmes hétérozygotes. Ceci explique sa prévalence élevée dans certains pays ou régions (Irlande, Nord du Royaume-Uni, Scandinavie, Bretagne, ...). On estime que pour l'ensemble de la population de la Scandinavie, la prévalence des sujets homozygotes est de 5 pour 1000.
Une deuxième mutation, H63D, est également connue et a une distribution géographique plus large. Les sujets homozygotes pour cette mutation, ou les sujets hétérozygotes composites (1 copie du gène avec la mutation C282Y, l'autre copie avec la mutation H63D) n'ont en général pas de surcharge en fer aussi sévère sauf s'il existe des facteurs associés, alcoolisme en particulier.
La protéine HFE fait partie des molécules HLA de classe I non classiques. Son rôle exact dans le métabolisme du fer n'est pas encore totalement compris. L'absence d'une protéine HFE fonctionnelle conduirait à un défaut de régulation de l'expression de certaines protéines impliquées dans le transport du fer ; tout se passerait comme si l'organisme concluait, à tort, à une carence en fer et tachait d'augmenter au maximum l'absorption digestive de celui-ci.
L'hémochromatose héréditaire mutation 845A (C282Y) dans le gène HFE a été décrite récemment, C282Y et les fréquences ont été signalées pour diverses populations mondiales. L'objectif d’une récente étude était de déterminer les fréquences de l'allèle Y de la mutation C282Y pour cinq populations françaises . La valeur plus élevée (= 5,6%) a été obtenue pour les Bretons, conformément à l'hypothèse indiquant une origine celtique de l'hémochromatose héréditaire de mutation.
Mode de transmission
La transmission est de type autosomique récessive : cela signifie que pour être malade il faut que les deux copies du gène HFE portent une mutation (on parle alors de sujet homozygote pour la mutation). La fréquence des hétérozygotes dans certaines régions d'Europe de l’Ouest explique la concentration des patients dans ces régions. Si les deux parents sont hétérozygotes, il y aura, statistiquement, un enfant sur quatre qui sera homozygote.
Les patients ayant la mutation à l'état homozygote (les 2 copies du gène sont mutées) peuvent développer une hémochromatose dans sa forme classique. Les patients hétérozygotes (1 seule copie du gène muté) n'ont pas de signe clinique mais peuvent transmettre la mutation à leur descendance. Cette mutation a une pénétrance assez faible : ainsi moins de la moitié des homozygotes ont une surcharge en fer et moins d'un tiers ont une atteinte clinique d'hémochromatose[2].
Description
L'hémocromatose est la première maladie génétique en France (en fréquence), avec une incidence estimée à 3/1000 naissances.
Les manifestations cliniques sont tardives, au-delà de 40 ans. Elles sont en effet liées au retentissement de la surcharge en fer sur certains tissus et organes et cette surcharge ne se met en place que lentement. Du fait des pertes menstruelles (règles) et des grossesses qui font perdre beaucoup de fer, la maladie s'exprime plus tardivement et moins sévèrement chez les femmes que chez les hommes.
Les principales manifestations cliniques sont :
- une fatigue (asthénie) inexpliquée ;
- troubles de la libido et ennuis sexuels ;
- un aspect « bronzé » qui est en fait une mélanodermie (coloration grise de la peau) ;
- des douleurs articulaires (signe de la poignée de main en particulier) révélant des arthrites ;
- une atteinte hépatique : simple élévation des transaminases, augmentation de volume du foie (hépatomégalie), cirrhose, carcinome hépato-cellulaire ;
- une atteinte cardiaque (insuffisance ventriculaire gauche, troubles du rythme) ;
- des atteintes endocriniennes : diabète, hypogonadisme hypogonadotrophique (pouvant être responsable d'une impuissance chez l'homme) et hypothyroïdie.
L'ensemble de ces manifestations ne s'observe qu'en cas de ferritine très élevée et sont réversibles sous traitement, sauf les atteintes hépatiques évoluées, et parfois, les douleurs abdominales. Seules les atteintes articulaires peuvent parfois s'aggraver malgré les saignées.
En raison des atteintes au foie et des risques accrus de cirrhose, cette maladie est prise en charge par les services Hépato-Gastro-Entérologie.
Diagnostic
Clinique
L'association des symptômes, quand ils sont tous présents est en soi évocatrice d'hémochromatose mais le diagnostic n'est pas toujours facile à évoquer cliniquement chez les sujets jeunes. La mélanodermie, discrète au début, est par exemple souvent interprétée comme une coloration normale de la peau par le patient compte tenu de son caractère ancien. D'autres symptômes (asthénie, douleurs articulaires) sont à la fois fréquents dans la population générale et non spécifiques.
Souvent ce sont en fait les anomalies biologiques qui feront évoquer le diagnostic.Biologique
Certaines anomalies non spécifiques, élévation des transaminases hépatiques en particulier, peuvent faire évoquer ce diagnostic.
L'étude du bilan martial va confirmer la surcharge en fer : augmentation de la concentration du fer sérique (dépassant 30 µmol/l) et du coefficient de saturation de la transferrine (supérieur à 45 % pouvant atteindre 80 à 100 %). La ferritine plasmatique est augmentée proportionnellement à l'intensité de la surcharge en fer de l'organisme. Le risque de survenue des complications viscérales est corrélé à l'importance du taux de ferritine.
Imagerie
L'Imagerie par résonance magnétique permet, d'une part, d'évaluer plus précisément la surcharge en fer des organes (notamment du foie) de façon moins invasive que la biopsie. D'autre part, une surveillance de pathologies secondaires à l'hémochromatose : hépatomégalie, cirrhose, carcinome hépatocellulaire et splénomégalie.
Différentiel
Avec les autres types d'hémochromatose :
Nom habituel Autres noms Fréquence Transmission Pénétrance Gène Chromosome Protéine en cause OMIM Hémochromatose génétique Hémochromatose de type 1 ou classique 1 sur 300 Récessive Basse HFE 6p21.3 HLA de classe I non classique 235200 Hémochromatose juvénile Hémochromatose de type 2A Rare Récessive HJV 1q21 Hémojuveline 602390 Hémochromatose juvénile Hémochromatose de type 2B Rare Récessive HAMP 19q13 Hepcidine 606464 Hémochromatose par mutation TFR2 Hémochromatose type 3 Très rare (moins de 20 cas connus) Récessive 100 % TFR2 7q22 Récepteur de la transferrine 2 604250 Hémochromatose type 4 Hémochromatose à transmission dominante Très rare Dominante SLC40A1 2q32 Ferroportine 606069 Génétique
Le diagnostic génétique est facile. Il se fait à partir d'un simple prélèvement sanguin. Les mutations C282Y et H63D sont recherchées directement par des techniques de biologie moléculaire et cet examen se fait maintenant en routine dans de nombreux laboratoires. En particulier, la recherche de la mutation C282Y s'effectue par une amplification de la séquence ADN du patient via une PCR. Ensuite l'enzyme RsaI va cliver la séquence en deux fragments si la personne est saine, et en trois fragments si la personne possède la mutation. On observe le résultat du clivage par électrophorèse sur gel d'acrylamide (de préférence pour avoir une bonne résolution). En cas de négativité du test, d'autres mutations, beaucoup plus rares, peuvent être recherchés.
Évolution
La plupart des sujets homozygotes pour la mutation C282Y ne présentent aucun signe clinique et n'évolueront pas vers la maladie en particulier si est mis en place, en cas de surcharge en fer significative, un programme de saignée.
Le risque d'atteinte du foie et de survenue d'un hépato carcinome est multiplié par quatre par rapport à une population sans hémochromatose[3] mais le risque absolu reste cependant faible[4].
Diagnostic différentiel
En dehors des causes secondaires, de diagnostic évident, il faudra, en l'absence de mutation du gène HFE, rechercher les mutations des autres gènes impliqués dans les hémochromatoses génétiques.
Une mutation du gène HFE peut venir aggraver une hémochromatose secondaire : accumulation du fer plus rapide, par exemple, chez un patient par ailleurs régulièrement transfusé.
Traitement
Il existe des recommandations sur la prise en charge de la maladie, dont les dernières, européennes, datent de 2010[5].
L'efficacité des traitements est grandement majorée par la découverte précoce de la maladie. Mis en place avant les symptômes graves, ils retardent voire empêchent leurs apparitions. Malheureusement une fois les symptômes hépatiques et rhumatismaux apparus, l'évolution est parfois moins favorable. Ils devront être pris en charge par d'autres traitements spécifiques des symptômes.
Le traitement principal est représenté par des saignées (phlébotomie) qui reste, à l'heure actuelle, le seul traitement véritablement efficace. On distingue « un traitement d'attaque » (saignées toutes les semaines ou tous les 15 jours jusqu'à normalisation du bilan martial), puis un « traitement d'entretien » (saignées régulières dont la périodicité sera adaptée à l'importance de la surcharge en fer. Le volume des saignées correspond habituellement à celui d'un don du sang (250 à 400 ml)). Le but est la normalisation de la ferritinémie et de la saturation de la transferrine afin d'éviter l'évolution vers les complications viscérales. Certains conseillent le maintien d'un taux bas de ferritine (inférieur à 20 μg/l). Les valeurs basses conseillées sont revues à la hausse depuis 2 ans (entre 2004 et 2006). Actuellement (2010), la valeur cible est une ferritinémie inférieure à 50 μg/l[5]. L'atteinte hépatique peut ainsi régresser sous ce type de traitement[6].
Les autres traitements ont une efficacité modérée mais représentent un complément ou une alternative en cas de mauvaise tolérance des saignées par le patient (anémie, mauvais capital veineux... ). Il consiste en l'absorption de chélateur du fer : La desferrioxamine[7] par injection sous-cutané. Cependant des précautions doivent être prises avant la prescription. Notamment : l'absence de grossesse et l'activité rénale par une surveillance de la créatinine. Il existe aussi des techniques basées sur l'érythraphérèse[8] (retrait uniquement des hématies et réinjection du plasma et des autres cellules) cependant l'appareillage nécessaire est très coûteux et peu d'établissement en dispose pour ce type de traitement.
Certains symptômes de la maladie se traduisent par des douleurs rhumatismales et abdominales, celles-ci peuvent être traitées par des antalgiques et anti-inflammatoires. Cependant, au vu des atteintes hépatiques potentielles ou avérées il est particulièrement important d'éviter les antalgiques hépato-toxique (tel que le paracétamol) et les glucocorticoïdes.
Un régime pauvre en fer est généralement recommandé mais l'efficacité réelle de ce type de régime n'a pas été évaluée[9].
Recherche
Des travaux de recherche[10] ont, en 2001, mis en évidence une hormone peptidique, sécrétée par le foie, qui inhibe l'absorption du fer alimentaire et son recyclage dans l'organisme en agissant sur les complexes ferroportines présents sur les entérocytes et les macrophages : L'hepcidine. La sécrétion de celle-ci est souvent altérée chez les patients atteint d'hémochromatose héréditaire.
Les tentatives de synthèse artificielle de l'hepcidine ont pour le moment échoué car la molécule (un peptide de 25 acides aminés) présente quatre ponts disulfure qui mettent en échec la plupart des techniques de synthèse en laboratoire.
Conseil génétique
La recherche systématique de la mutation n'est pas recommandée, la pénétrance de la maladie restant faible.
Quand une hémochromatose est diagnostiquée chez un patient, un bilan martial (dosage du fer sérique, de la transferrine et de la ferritine) est proposé chez les apparentés au premier degrè (parents, frères, sœurs, et enfants du patient) et peut être étendu éventuellement aux autres membres de la famille. La recherche de la mutation n'est proposé qu'en cas d'anomalie de ce bilan martial[9].
Compte tenu de la bénignité de la maladie quand elle est prise en charge, un diagnostic prénatal (jamais dénué de risque) n'est pas justifié.
Voir aussi
Articles connexes
Notes et références
- (en) J.N. Feder, A. Gnirke, W. Thomas, Z. Tsuchihashi, D.A. Ruddy et A. Basava, « A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis », dans Nature Genetics, no 13, 1996, p. 399-408
- Whitlock E, Garlitz B, Harris E, Beil T, Smith P, Screening for hereditary hemochromatosis: a systematic review for the US Preventive Services Task Force, Ann Intern Med, 2006;145:209-23
- Ellervik C, Birgens H, Tybjærg-Hansen A, Nordestgaard BG, Hemochromatosis genotypes and risk of 31 disease endpoints: meta-analyses including 66 000 cases and 226 000 controls, Hepatology, 2007;46:1071-80.
- Asberg A, Hveem K, Thorstensen K et Als. Screening for hemochromatosis—high prevalence and low morbidity in an unselected population of 65 238 persons, Scand J Gastroenterol, 2001;36:1108-15
- European Association for the Study of the Liver, EASL clinical practice guidelines for HFE hemochromatosis, J Hepatol, 2010;53:3-22
- Falize L, Guillygomarc’h A, Perrin M et Als. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases, Hepatology, 2006;44:472-7
- biam2.org, « Desferrioxamine mésilate »
- Rombout-Sestrienkova E, van Noord PA, van Deursen CT et Als. Therapeutic erythrocytapheresis versus phlebotomy in the initial treatment of hereditary hemochromatosis—a pilot study, Transfus Apher Sci, 2007;36:261-7
- van Bokhoven MA, van Deursen CTBM, Swinkels DW, Diagnosis and management of hereditary haemochromatosis, BMJ, 2011;342:c7251
- Hepcidine - Le métabolisme du fer et ses maladies
Bibliographie
- (en) R. Saddi et J. Feingold, « Idiopathic haemochromatosis : An autosomal recessive disease », dans Clin Genet, no 5, 1974, p. 234-241
- (en) M. Simon, M. Bourel, B. Genetet et R. Fauchet, « Idiopathic haemochromatosis. Demonstration of recessive inheritance and early detection by family HLA typing », dans New England Journal of Medicine, no 297, 1977, p. 1017-1021
- M. Simon, Y. Pawlotsky, M. Bourel, R. Fauchet et B. Genetet, « Hémochromatose idiopathique : maladie associée à l'antigène tissulaire HLA-A3 », dans Nouvelles Presses Médicales, no 4, 1975, p. 1432
- (en) M. Simon, L. Le Mignon, R. Fauchet, J. Yaouanq, V. David et Edan, « A study of 609 haplotypes marking for the hemochromatosis gene : (1) mapping of the gene near the HLA-A locus and characters required to define a heterozygous population and (2) hypothesis concerning the underlying cause of hemochromatosis-HLA association », dans American Journal of Human Genetics, no 41, 1987, p. 89-105
- (en) E.C. Jazwinska, S.C. Lee, S.I. Webb, J.W. Halliday et L.W. Powell, « Localization of the hemochromatosis gene close to D6S105 », dans American Journal of Human Genetics, no 53, 1993, p. 347-352
- (en) G. Gandon, A.M. Jouanolle, B. Chauvel, V. Mauvieux-Lelaure, Le Treut et J. Feingold, « Linkage disequilibrium and extended haplotypes in the HLA-A - D6S105 region : implications for mapping the haemochromatosis gene (HFE) », dans Human Genetics, no 97, 1996, p. 103-113
- (en) A.M. Jouanolle, G. Gandon, R. Jézéquel, M. Blayau, M.L. Campion et J. Yaouanq, « Haemochromatosis and HLA-H », dans Nature Genetics, no 14, 1996, p. 251-252
- (en) M. De Sousa, R. Reimao, R. Lacerda, P. Hugo, S.H.E. Kaufmann et G. Porto, « Iron overload in beta2 microglobulin deficient mice », dans Immunology Letters, no 39, 1994, p. 105-111
- (en) B.E. Rothenberg et Y.R. Voland, « Beta2 knock out mice develop parenchymal iron overload : a putative role for class I genes of the major histocompatibility complex in iron metabolism », dans Proceedings of the National Academy of Sciences of the USA, no 93, 1996, p. 1529-1534
- M. Vernet, B. Poggi, F. Bienvenu, M.C. Carlier, C. Chapuis-Cellier et C. Fleuret, « Incidence de la standardisation du dosage de la transferrine sérique par le CRM 470 sur le coefficient de saturation en fer de la transferrine », dans Annales de biologie clinique, no 54, 1996, p. 171-175
- C. Guillemin, M.C. Revenant, M. Vernet, J.F. Dezier, C. Charlier, M. Paris et M. Pressac, « La ferritine érythrocytaire », dans Annales de biologie clinique, no 51, 1993, p. 605-609
- (en) Y. Gandon, D. Guyader, J.F. Heautot, M.I. Reda, J. Yaouanq et T. Buhe, « Hemochromatosis : diagnosis and quantification of liver iron with gradient-echo MR imaging », dans Radiology, no 193, 1994, p. 533-538
- (fr) Site en français de renseignement sur les maladies rares et les médicaments orphelins
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:235200[1]
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 [2]
Liens externes
- Association Hemochromatose France
- Fédération Française des Associations de Malades de l'Hémochromatose
- (fr) Centre de référence français des surcharges en fer rares d'origine génétique
Catégories :- Maladie métabolique congénitale
- Maladie génétique du métabolisme des métaux
- Maladie génétique en hématologie
Wikimedia Foundation. 2010.