- Sequencage des proteines
-
Séquençage des protéines
Le séquençage des protéines est la détermination de la séquence polypeptidique. Elle est destinée à connaître le nombre, la nature chimique et l'ordre de tous les résidus d'acides aminés dans un polypeptide. Pour cela, si la protéine contient plus d'une chaîne polypeptidique, les chaînes doivent être d'abord séparées, puis purifiées. Généralement, toutes les liaisons disulfures seront réduites et les thiols ainsi obtenus alkylés. Après isolement d'une chaîne polypeptidique, la séquence peut être étudiée:
- en faisant appel à des méthodes récurrentes chimiques ou enzymatiques libérant les acides aminés un à un à partir de l'extrémité C ou N terminale;
- ou plus récemment par des méthodes de spectrométrie de masse (séquençage de novo).
Le séquencage des protéines est plus difficile que celui de l'ADN.
Sommaire
- 1 Historique
- 2 Les structures
- 3 La dénaturation
- 4 Détermination de la composition d'une protéine en acides aminés
- 5 Détermination de la séquence
- 6 Notes et références
- 7 Références
Historique
C'est dans les années 50 que Frederick Sanger et les chercheurs de l'Université de Cambridge ont déterminé la séquence des acides aminés qui composent l'insuline. Pour cela, plutôt que d'hydrolyser complètement l'hormone, ils ont utilisé des enzymes catalysant la coupure des polypeptides au niveau de sites spécifiques. Les fragments résultants ont été séparés par chromatographie, puis partiellement hydrolysés pour donner un nouveau groupe de fragments. À ce stade, Sanger utilisa des méthodes chimiques pour déterminer la séquence des acides aminés. À partir des séquences obtenues il rechercha des chevauchements afin de reproduire la séquence initiale. Il fallut plusieurs années à Sanger pour reconstituer la structure primaire de l'insuline. Depuis, la plupart des étapes du séquençage des protéines ont été automatisées. Actuellement le séquençage des protéines a été largement délaissé au profit de celui des acides nucléiques (ADN principalement et ARN), plus rapide et plus performant.
Les structures
Chaque protéine est une macromolécule constituée par l'enchaînement de molécules de masse moléculaire plus petite appelées monomères : les acides aminés.
La structure primaire
Les acides aminés (AA) s'assemblent grâce à des liaisons peptidiques (groupement cétone d'un AA avec le groupement amine d'un AA voisin → C=O—NH2), et forment ainsi la structure primaire.
La structure secondaire
La structure secondaire peut se manifester sous deux formes:
- Élément A : L'hélice α (en spirale – exemple : la kératine). Ce sont les protéines fibreuses.
- Élément B : Le feuillet plissé β (en escalier – exemple : l'hémoglobine). Ce sont les protéines globulaires.
Cette structure est due à l'existence de liaisons hydrogène qui se forment entre des liaisons peptidiques voisines.
La structure tertiaire
Soit le feuillet plissé β, soit l'hélice α s'assemblent en structure tertiaire[1] par des liaisons secondaires qui se créent entre les radicaux des acides aminés qui, lors de la formation de la structure secondaire, se sont retrouvés à l'extérieur de la structure (exemples : liaison saline, hydrogène, pont disulfure). Ces liaisons, hormis le pont disulfure qui est une liaison covalente forte, sont des liaisons covalentes faibles.
Ce repliement de la chaîne peptidique amène la création d'un site actif responsable de l'activité biologique de la protéine.
La structure quaternaire
Certaines protéines, dites oligomériques (constituées par plusieurs chaînes de polypeptides – chaque chaîne présente une structure primaire, secondaire et tertiaire), telle que l'hémoglobine par exemple, atteignent une structure quaternaire en adoptant une construction symétrique et paire par l'association de ces différents protomères. Ces protéines ne voient la création de leur site actif que lorsque cette structure a été atteinte.
La dénaturation
La dénaturation est une désorganisation de la structure spatiale sans rupture des liaisons peptidiques, car seules les liaisons secondaires sont concernées.
Conséquences de la dénaturation
- Perte de l'activité biologique
- Changement des propriétés optiques
- Perte de la solubilité car précipitation
Les agents dénaturants
Ils sont nombreux et peuvent être physiques ou chimiques.
Les agents dénaturants physiques
- Température : Toute augmentation de la température provoque une rupture des liaisons hydrogène.
- Modification de pH : Elle entraîne une modification des charges portées par les groupements ionisés, d'où une rupture de ces liaisons.
Les agents dénaturants chimiques
- L'urée et le SDS (détergent) : Rupture les liaisons hydrogène.
- Le β mercaptoéthanol et l'acide performique : Rupture des ponts disulfures.
- Les bases et détergents provoquent une forte dénaturation protéique.
- Les défécants : Précipitation des protéines (défécation)
- Le bromure de cyanogène (BrCN) : clive la méthionine.
Importance de la dénaturation
In vitro : pour les extractions d'enzymes, on évite les dénaturations. Mais pour une stérilisation, on les provoque.
C'est également en faisant agir des dénaturants sur des protéines que l'on va pouvoir les séquencer.
Détermination de la composition d'une protéine en acides aminés
Rupture des ponts disulfure
La rupture des ponts disulfure entre les cystéines est avant tout une dénaturation chimique, qui est de plus essentielle pour pouvoir séparer les différents groupes d'acides aminés. On utilise à cet effet des agents réducteurs porteurs de fonctions thiol (–SH), comme le 2-mercaptoéthanol ou le dithiothréitol (réactif de Cleland). Il existe également des réducteurs non-thiolés comme le TCEP (Tris (2-carboxyethyl) phosphine). Il faut savoir également que deux cystéines sont réunies par leur groupement thiol (–SH), et forment alors une molécule appelée cystine. La cystéine est un acide aminé, elle peut donc être dénaturée. Le pont disulfure est responsable de la structure tertiaire des protéines, apportant une grande stabilité a cette dernière.
Hydrolyse des liaisons peptidiques
L'hydrolyse des liaisons peptidiques permet de séparer les différents acides aminés, car elles sont responsables de la structure primaire, agissant entre le groupement cétone d'un acide aminé (C=O) et le groupement amine (NH2) d'un AA (acide aminé) voisin.
- Hydrolyse totale acide :
- Utilisation d'acide chlorhydrique (HCl) à 6 mol/L à chaud (110°C) et pendant 24h environ.
- L'hydrolyse acide totale détruit le tryptophane et transforme en acide les fonctions amides de la glutamine et de l'asparagine pour donner du glutamate et de l'aspartate.
- Hydrolyse totale alcaline:
- Utilisation d'hydroxyde de sodium (NaOH) à 4 mol/L à chaud (110°C) pendant 24h environ.
- L'hydrolyse totale alcaline détruit la sérine, la thréonine, la cystéine.
L'hydrolyse totale acide détruisant moins d'acides aminés, elle est en conséquence la méthode d'hydrolyse la plus utilisée.
Analyse de l'hydrolysat
Après rupture des liaisons disulfures au 2-mercaptoéthanol et une hydrolyse, l'hydrolysat est déposé sur une résine échangeuse d'anions ou de cations. Une fois les fractions récupérées on les colore à la ninhydrine et on mesure l'absorbance à 570nm. On obtient finalement une estimation du pourcentage de chaque acide aminé contenu dans la chaîne (% de résidus alanine,...). La détermination de cette composition n’est pas nécessaire pour déterminer la séquence et est de fait de moins en moins pratiquée.
Détermination de la séquence
Le marquage chimique de l'extrémité N-terminal (NH2)
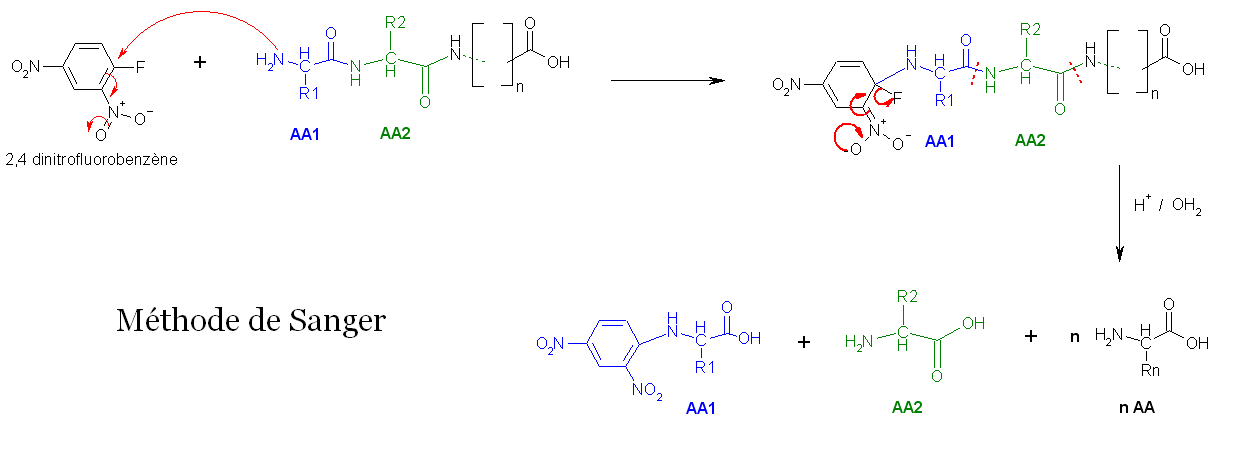
- La première méthode utilise la réaction de Sanger (cf. arylation). Les fonctions 2-amine des acides aminés et peptides réagissent avec le 2,4-dinitrofluorobenzène pour former des dérivés jaunes, les 2,4-dinitrophényl-peptides. Lorsque ces derniers composés sont soumis à une hydrolyse acide, toutes les liaisons peptidiques sont hydrolysées, mais la liaison entre la fonction 2,4-dinitrophényl et la fonction amine de l'acide aminé N-terminal restent stables, d'où l'identification de ce dernier.
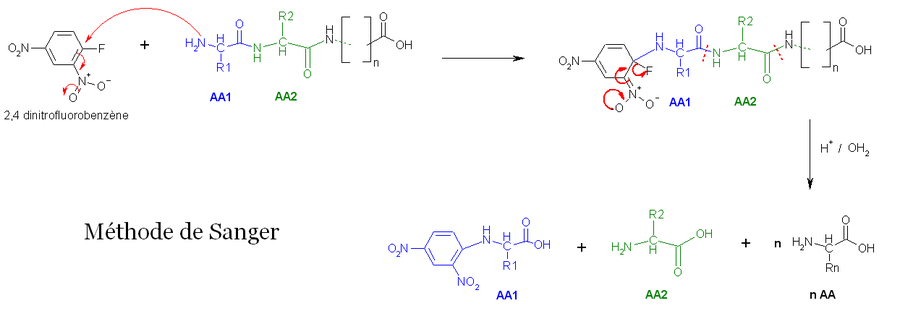
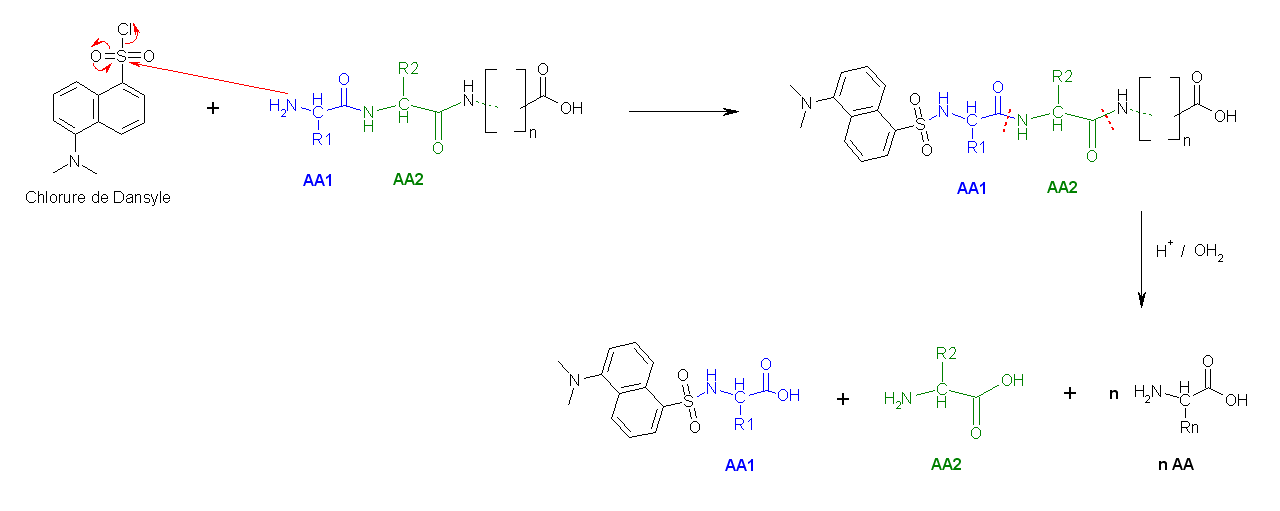
- La deuxième méthode utilise le chlorure de dansyle, qui, combiné à une hydrolyse totale acide forme un Dansyl-AcideAminé1 (DNS-aa) qui est fluorescent et un hydrolysat.
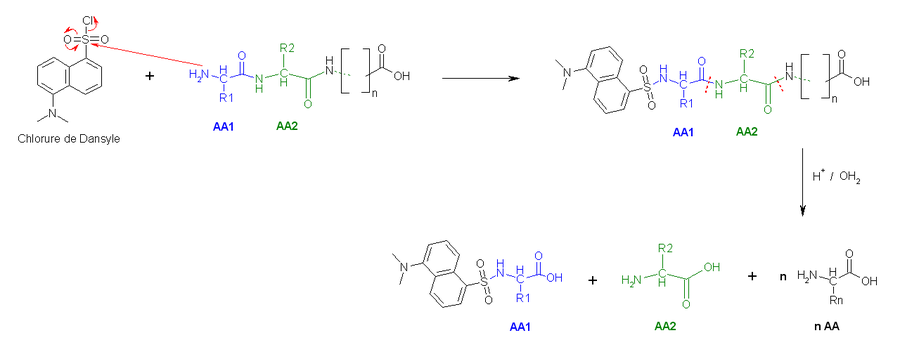
- Une troisième méthode est celle d'Edman, dans laquelle le phénylisothiocyanate (PITC) réagit avec la fonction amine N-terminale pour donner un complexe qui, après action d'un acide dans des conditions douces libère une phénylthiohydantoïne et le peptide restant intact (cf. carbamylation). Les deux premières méthodes utilisant une hydrolyse totale acide, le reste de la chaîne peptidique voit ses acides aminés clivés. Le grand avantage de la méthode d'Edman est que la chaîne peptidique reste intacte et peut être récupérée et soumise à nouveau au traitement avec le PITC.
Identification des acides aminés C-terminaux
On utilise l'hydrazynolise, qui coupe les liaisons à 100°C et donne un acide aminé libre. On peut également utiliser des carboxypeptidases qui coupent l'extrémité C-terminale, il en existe plusieurs types:
- la carboxypeptidase A, qui coupe tout sauf les acides aminés basiques (elle est bloquée par la proline)
- la carboxypeptidase B, qui est spécifique des acides aminés basiques (Lys, Arg)
Analyse enzymatique des extrémités N et C-terminaux
Il existe également des méthodes biochimiques utilisant des enzymes capable de couper le premier ou le dernier acide aminé de la chaîne polypeptidique, on les appelle des exopeptidases. Les aminopeptidases clivent la liaison peptidique située juste après le premier acide aminé et libèrent celui-ci. De manière symétrique, les carboxypeptidases clivent la liaison peptidique située juste avant le dernier acide aminé.
Il en existe plusieurs de ces enzymes de spécificités variées, par exemple parmi les carboxypeptidases, on trouve :
- La carboxypeptidase A, clive l'acide aminé C-terminal lorsque celui-ci est aromatique ou aliphatique
- La carboxypeptidase B, clive l'acide aminé C-terminal lorsque celui-ci est basique (Arginine ou Lysine)
- La carboxypeptidase Y à spectre large
En analysant les acides aminés libérés par ces enzymes, il est possible d'analyser la séquence N ou C-terminale de la protéine.
Fragmentation de la chaîne polypeptidique
La chaîne polypeptidique est découpée en une série de petits peptides par des hydrolyses enzymatiques ou chimiques.
Méthode chimique
L'hydrolyse chimique la plus utilisée implique une réaction avec le bromure de cyanogène, qui scinde les liaisons peptidiques dans lesquelles la fonction carboxyle est fournie par des résidus de méthionine.
Méthode enzymatique (utilisation d'endopeptidases)
L'hydrolyse enzymatique utilise les protéases, enzymes qui hydrolysent les liaisons peptidiques. La trypsine catalyse l'hydrolyse des liaisons peptidiques dans lesquelles la fonction carboxyle est fournie par la lysine ou l'arginine, la chymotrypsine permet la coupure du côté C-terminal des acides aminés aromatiques (Phe, Trp et Tyr).
- Trypsine (C) : LYS, ARG (sauf si PRO à droite)
- Chymotrypsine (C) : PHE, TRP, TYR (sauf si PRO à droite)
- Thermolysine (N) : LEU, ILE, VAL (sauf si PRO à gauche)
- Pepsine (N) : PHE, TRP, TYR (sauf si PRO à gauche)
- LysC
- Papaïne : très utilisée dans l’industrie agro-alimentaire et chimique, elle permet notamment en boucherie d’attendrir la viande. Elle est aussi utilisée dans les produits de nettoyages de verres de contact, ainsi que dans les poudres à lessive, où son action est de détruire les protéines responsables des taches.
- Clostripaïne
- Subtilisine
- Elastase
- etc.
Identification de la séquence
L'identification de la séquence se fait de manière logique à partir des résultats obtenus par l'utilisation des différentes méthodes décrites précédemment.
Séquençage par spectrométrie de masse
La fragmentation par spectrométrie de masse MS/MS permet de séquencer des courtes séquences d'acides aminés (10 à 20). Couplée avec les techniques de digestion des protéines précédemment décrite, il est ainsi possible de connaitre la séquence d'une protéine. Cependant, cette approche nécessite d'utiliser différents protocoles de digestion différents pour espérer obtenir l'ensemble de la séquence de la protéine.
Pour un organisme dont le génome est entièrement connu, on utilisera un seul type de digestion (par exemple une digestion par la trypsine suivie d'une analyse par HPLC couplée à un spectromètre de masse de type ESI-MS/MS), ce qui permet d'obtenir jusque 30% de la séquence de la protéine (ce qui est suffisant pour caractériser la protéine).
Pour un organisme dont le génome n'est pas entièrement connu, on utilise en première intention un séquencage automatisé (séquençace d'Edman par exemple).
Notes et références
- Cours de première année de Biochimie
Références
 Portail de la biochimie
Portail de la biochimie
Catégories : Méthode de la biochimie | Protéomique
Wikimedia Foundation. 2010.