- Chromatographie
-
La chromatographie est une technique physique de séparation d'espèces chimiques. L'échantillon contenant une ou plusieurs espèces est entraîné par un courant de phase mobile (liquide, gaz ou fluide supercritique) le long d'une phase stationnaire (papier, gélatine, silice, polymère, silice greffée etc) ; chaque espèce se déplace à une vitesse propre dépendant de ses caractéristiques et de celles des deux phases.
Cette technique d'analyse chimique peut être couplée à un détecteur en vue d'une analyse qualitative ou quantitative du milieu.
La chromatographie analytique est utilisée pour identifier ou doser les composés chimiques d'un mélange et apprécier leur concentration.
La chromatographie préparative est utilisée pour purifier assez de produit pour d'autres utilisations. Son but est d'obtenir de la substance ; c'est pourquoi, à toute échelle, elle implique de collecter des fractions.
Sommaire
Définitions
Chromatographie : méthode physique de séparation basée sur les différences d'affinités des substances à analyser à l'égard de deux phases, l'une stationnaire ou fixe, l'autre mobile. Selon la technique chromatographique mise en jeu, la séparation des composants entraînés par la phase mobile, résulte soit de leur adsorption et de leur désorption successives sur la phase stationnaire, soit de leur solubilité différente dans chaque phase.
Phase stationnaire : phase fixe soit sur la surface intérieure d'une colonne soit sur une surface plane.
Phase mobile : phase qui se déplace à travers la phase stationnaire, en entraînant les analytes. La phase mobile ne doit pas interagir avec la phase stationnaire mais uniquement avec les analytes.
Chromatogramme : graphique d’une fonction de la concentration en analyte en fonction du temps (ou du volume) d’élution.
Étymologie
Le mot chromatographie vient du grec ancien Khrôma qui signifie "couleur" et Graphein qui signifie "écrire"
Histoire
Le botaniste russe Mikhail Tswett (1872-1919) fut, en 1906[1], le premier à utiliser le terme chromatographie.
À partir de 1903, Tswett utilisa des colonnes d'adsorption pour séparer des pigments de plantes. On spécula donc l'étymologie du mot « chromatographie » à partir du grec khrôma- pour couleur et donc pigment. Toutefois, Tswett ne donna jamais cette explication, mais tswett est le mot russe pour « couleur ».
En 1952, Martin et Synge reçurent le prix Nobel de chimie pour leur invention de la chromatographie de partage[2].
Principe
La chromatographie repose sur l'entraînement d'un échantillon dissous par une phase mobile à travers une phase stationnaire. Celle-ci retient plus ou moins fortement les substances contenues dans l'échantillon dilué selon l'intensité des forces d'interactions de faible énergie (comme les forces de Van der Waals, les liaisons hydrogène, etc.) réalisées entre les différentes espèces moléculaires et la phase stationnaire.
Les différents composants de l'échantillon ont généralement une vitesse caractéristique qui permet de les séparer, voire de les identifier. Cette vitesse de séparation est fortement dépendante de la nature de la phase mobile et de la phase stationnaire.
Souvent, l'échantillon est analysé par comparaison avec des substances déjà connues dans l'échantillon ou par comparaison avec les résultats de l'analyse d'une solution-étalon (solution commerciale contenant des substances connues, à des concentrations bien connues). Ces substances servent de références et permettent d'identifier ou de doser chaque espèce par comparaison des vitesses de séparation (et éventuellement d'autres renseignements donnés par la détection). Il s'agit de chromatographie analytique.
Dans d'autres cas, on se contente de séparer les fractions, de les récolter pour les identifier par d'autres techniques : c'est la chromatographie préparative.
Il existe de nombreux types de chromatographie ; on peut notamment les classer selon la nature de la phase mobile :
- la chromatographie sur couche mince (CCM ou TLC en anglais) ;
- la chromatographie en phase gazeuse (CPG ou GC en anglais) également appelée CPV (chromatographie en phase vapeur) ;
- la chromatographie en phase liquide (CPL ou LC en anglais) ;
- la chromatographie en phase liquide à haute performance (CLHP ou HPLC en anglais) ;
- la chromatographie en phase supercritique (CPS ou SFC en anglais).
On peut aussi les nommer selon les interactions développées par la phase stationnaire :
- la chromatographie d'adsorption/d'affinité ;
- la chromatographie de partage ;
- la chromatographie à échange d'ions ;
- la chromatographie chirale (qui est, soit de la CPG, soit de la CPL) ;
- la chromatographie d'exclusion stérique (CES ou SEC en anglais) ;
Ou selon le support de la phase stationnaire :
- la chromatographie sur colonne (regroupant notamment HPLC et CPG) : la phase stationnaire est dans un tube étroit et la phase mobile progresse par gravité ou différence de pression ;
- la chromatographie planaire[3] (qui recouvre CCM et chromatographie sur papier) : la phase stationnaire est sur la surface d'un support plat (CCM) ou dans une feuille de cellulose poreuse (chromatographie papier) et la phase mobile se déplace par capillarité ou par gravité.
Enfin, un nouveau type de chromatographie commence à trouver des applications : la chromatographie à 2 dimensions.
Exemple : la chromatographie sur papier
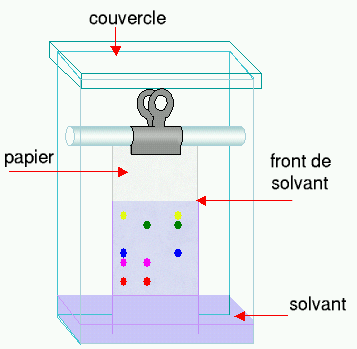
 schéma d'une chromatographie sur papier
schéma d'une chromatographie sur papier
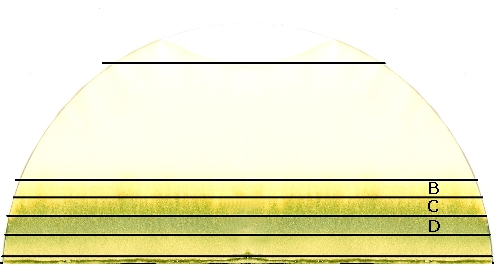
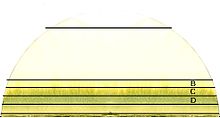 Résultat d'une chromatographie de base sur papier filtre. Fait avec des feuilles d'épinard
Résultat d'une chromatographie de base sur papier filtre. Fait avec des feuilles d'épinardLa chromatographie sur papier est une technique de chromatographie en phase liquide.
Pour effectuer une telle séparation, une petite quantité de la ou des solutions à analyser est déposée sur le bord d'une bande de papier de chromatographie. Cet échantillon est adsorbé par le papier ; ce qui signifie que les molécules interagissent avec ce dernier et qu'elles auront tendance à rester au même endroit.
Le papier est ensuite trempé dans un solvant (éluant) comme un mélange eau/éthanol et placé dans un récipient fermé. Pendant que le solvant (éluant) monte le long du papier par capillarité, il rencontre l'échantillon et l'entraîne.Les différentes substances constituant l'échantillon migrent à différentes vitesses selon qu'elles interagissent plus ou moins fortement avec le papier.
La chromatographie sur papier demande un certain temps (généralement plusieurs heures). Une fois l'opération terminée, généralement quand le front de solvant (éluant) est presque arrivé en haut du papier, le papier est retiré de la cuve et on laisse évaporer le solvant. Le résultat est appelé chromatogramme.
Le chromatogramme est utilisé pour comparaison avec d'autres analyses effectuées sur des substances connues et prises dans des conditions identiques, pour identifier les substances de l'échantillon. Les substances peuvent être identifiées en calculant la valeur Rf qui peut être comparée à celles se trouvant dans les tables. Cette valeur est calculée de la façon suivante : Rf = (distance parcourue par l'échantillon) / (distance parcourue par le solvant)
Il y a plusieurs façons d'identifier les endroits où se trouvent les produits ainsi séparés :
- les produits sont colorés, il n'y a rien de spécial à faire.
- les produits sont fluorescents, on peut les identifier sous une lampe ultraviolette.
- sinon, il faudra utiliser un révélateur qui réagira chimiquement avec les produits (en les détruisant) et dont le résultat sera coloré.
Pour séparer des mélanges complexes de substances similaires, il peut être utile d'employer la chromatographie à deux dimensions. Celle-ci s'effectue en deux étapes entre lesquelles on change de solvant et on tourne le papier de 90°. Les interactions développées par le nouveau solvant seront différentes, ce qui modifiera la séparation dans cette deuxième dimension et permettra une meilleure séparation globale.
La chromatographie sur papier peut aussi constituer une technique micro-préparative : pour récupérer les produits ainsi séparés, les portions du papier où ils se situent sont découpées et redissoutes ensuite. Les quantités récupérables sont de l'ordre du milligramme ou moins.
Étapes d'une analyse quantitative
- Choix de la méthode
- Analytes à étudier : nature et nombre
- Nombre d’analyses
- Exactitude recherchée
- Échantillonnage
- Préparation de l’échantillon
- Mise en solution
- Extraction des analytes de l’échantillon
- Concentration
- Rendement de l’extraction
- Éliminer les interférences
- Effet de matrice
- Purification de l’extrait
- Analyse chromatographique
- Directe
- Après traitement (méthylation, sylilation…)
- Étalonnage
- Linéarité
- Calcul des résultats
- Exactitude
- Evaluation d’incertitude
Grandeurs caractéristiques en analyse chromatographique sur colonne
Vitesses de déplacement des solutés
Le pouvoir séparateur d’une colonne est fonction des vitesses relatives d’élution. Ces vitesses sont donc fonction du coefficient de distribution des solutés entre les 2 phases :

Coefficient de distribution :

où CS est la concentration du soluté en phase stationnaire et CM la concentration du soluté en phase mobile.
Temps de rétention tR : temps qui s’écoule entre l’injection de l’échantillon et l’apparition d’un pic de soluté sur le détecteur d’une colonne chromatographique.
Temps mort tM : temps nécessaire pour qu’une espèce non retenue par la phase stationnaire traverse la colonne.
Vitesse linéaire moyenne de déplacement du soluté :

où L est la longueur de la colonne.
Vitesse linéaire moyenne de la phase mobile :

Relation entre vitesse de déplacement et coefficient de distribution :

où VS et VM sont les volumes de solutés respectivement dans la phase stationnaire et mobile (aussi appelé volume mort), or ces volumes sont proportionnels aux volumes des 2 phases respectivement, ils peuvent donc être estimés si on connaît la structure géométrique de la colonne.
Temps réduit t'R :
t'R = tR − tM Volume réduit V'R :
V'R = VR − VM Débit D :

Certains paramètres qui affectent les vitesses relatives des analytes peuvent être modifiés :
- Vitesse linéaire de la phase mobile

- Coefficient de diffusion dans la phase mobile
- Coefficient de diffusion dans la phase stationnaire
- Facteur de capacité k’
- Diamètre des particules support
- Epaisseur de la couche liquide sur la phase stationnaire
Facteur de capacité
Facteur de capacité k' : paramètre expérimental important permettant de décrire la vitesse de progression des solutés dans les colonnes. C’est le rapport des quantités d’un analyte présentes à l’équilibre dans les 2 volumes de phase stationnaire et mobile adjacentes. Le facteur de capacité depend de la température (CPG), du remplissage de la colonne (CPG), de la composition de la phase mobile et stationnaire (HPLC)...

- k'<1 : élution trop rapide
- 1<k'<5 : élution optimale
- 5<k' : élution trop lente
Facteur de sélectivité α : rapport du coefficient de distribution du soluté qui est retenu le plus sur le coefficient de distribution du soluté qui est retenu le moins. Le facteur de sélectivité de deux analytes permet d’estimer à quel point la colonne peut les séparer. Il est donc utilisé pour calculer le pouvoir séparateur d’une colonne. α>1 toujours.

Élargissement des bandes et efficacité d'une colonne
L’efficacité d’une colonne dépend du degré d’élargissement du pic qui se produit à mesure que l’analyte parcourt la colonne. Cet élargissement dépend du temps de séjour de l’analyte dans les 2 phases :
- tR grand : pics larges ;
- tR petit : pics fins.
L’efficacité d’une colonne s’évalue à partir de l’un ou l’autre de ces deux termes :
- H = hauteur équivalente à un plateau théorique
- N = nombre de plateaux théoriques
L’efficacité augmente quand N augmente ou quand H diminue à L constante. σ est la variance, ω est la largeur de la base du pic, δ la largeur du pic à mi-hauteur :
 et
et 
 ou
ou 
L’importance des effets cinétiques sur l’efficacité de la colonne dépend essentiellement de la durée de contact entre la phase mobile et la phase stationnaire, dépendant elle-même de la vitesse d’écoulement de la phase mobile. L’élargissement des pics est minimisé en réduisant la granulométrie du matériau de remplissage et le diamètre de la colonne. En effet la phase stationnaire est granulée et le solvant occupe le volume des interstices, ce volume est appelé volume mort de solvant. H augmente avec ce volume et diminue avec le diamètre de la colonne.
Résolution de la colonne
La résolution RS d'une colonne est la mesure quantitative de son aptitude à séparer deux analytes A et B :

- Si RS > 1,5 : séparation complète de A et B
- Si
 : séparation incomplète de A et B
: séparation incomplète de A et B - Si RS < 0,75 : analytes A et B mal séparés
Remarque : une résolution de 1,5 correspond à un recouvrement des pics de 1%.
Lien entre résolution et nombre de plateaux théoriques :
![R_S=\frac{1}{4}\sqrt[2]{N}\left(1-\alpha\right)\frac{k'}{1-k'}](0/0b0aa7430a09ee57efe961f412c5937a.png)
Cette relation permet de remarquer que la résolution RS d'une colonne est proportionnelle à la racine carrée de sa longueur car le nombre de pics théoriques N est proportionnel à la longueur L de la colonne. Avec une phase stationnaire donnée on peut améliorer la résolution de la colonne en augmentant le nombre de plateaux théoriques donc en augmentant la longueur de la colonne. Mais ceci augmente la durée de l’analyse, un compromis est donc nécessaire.
Méthodes d'optimisation
- Modification de la hauteur équivalent à un plateau théorique (diamètre colonne, granulométrie…)
- Modification du facteur de capacité (composition phase mobile, température, solvant…)
- Modification du facteur de sélectivité (composition phase mobile, température colonne, composition phase stationnaire, effets chimiques (complexes…)
Notes et références
- Mikhail Tswett, « Adsorption analysis and chromatographic method. Application on the chemistry of the Chloropyhlls. », dans Berichte der Deutschen Botanischen Gesellschaft, vol. 24, 1906, p. 384-393
- Chemistry 1952
- IUPAC Compendium of Chemical Terminology, Electronic version, http://goldbook.iupac.org/P04682.html
Voir aussi
- Diffusion de la matière
- Migration (matière)
- Chromatographie en phase gazeuse
- Chromatographie en phase liquide
- Chromatographie en phase supercritique
- Chromatographie chirale
- Chromatographie en phase liquide à haute performance
- Chromatographie d'exclusion stérique
- Chromatographie de partage
- Chromatographie à échange d'ions
Liens externes
- Cours de chromatographie (ne comprend pas l'HPLC)
- Site du gouvernement
Wikimedia Foundation. 2010.