- Chorée de Huntington
-
 Pour les articles homonymes, voir Huntington.
Pour les articles homonymes, voir Huntington.Chorée de Huntington
Classification et ressources externes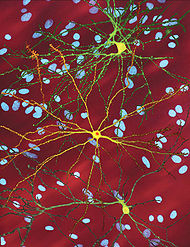
Image au microscope présentant la Chorée de Huntington. CIM-10 G10, F02.2 CIM-9 333.4, 294.1 OMIM 143100 DiseasesDB 6060 MedlinePlus 000770 eMedicine article/1150165 article/792600 MeSH D006816 La chorée de Huntington, aussi connue sous le nom de maladie de Huntington, est une maladie héréditaire ou de naissance à évolution probable vers la mort. Il existe cependant des traitements expérimentaux.
Cette maladie ne se développe, le plus souvent, que chez les personnes âgées de 40 à 50 ans. Toutefois il est possible que les symptômes apparaissent chez des patients plus jeunes : chez certains individus, les premiers symptômes apparaissent entre 15 et 25 ans. Elle se traduit par une dégénérescence neuronale affectant les fonctions motrices et cognitives aboutissant à une démence. Ses manifestations psychologiques s'accompagnent de manifestations neurologiques avec, chez le sujet éveillé, des gestes incohérents et anormaux (mouvements choréiques) indépendants de sa volonté, des troubles de l'équilibre et une léthargie.
La chorée de Huntington est souvent confondue avec la danse de Saint-Guy, qui est en fait la manifestation d'une chorée, quelle qu'en soit la cause.
Sommaire
Historique
La maladie a été décrite dès le XIXe siècle et son origine génétique suspectée. George Huntington en fit la description la plus complète en 1872, donnant son nom à cette affection[1].
Causes
Le gène Huntingtin fournit normalement l'information génétique pour synthétiser une protéine, la « huntingtine ». La mutation du gène provoque la synthèse d'une autre forme de protéine, dont la présence entraîne des dommages progressivement croissants à des zones spécifiques du cerveau. Le processus exact n'est pas entièrement connu. Les tests génétiques peuvent être effectués à tout stade de développement, avant même l'apparition des symptômes, soulevant des débats éthiques.
Il s'agit d'une mutation du gène IT15 situé sur le locus p16.3 du Chromosome 4. Cette mutation consiste en une expansion ou répétition de la séquence du trinucléotide CAG ou extension polyglutamique. Il s'agit donc d'un microsatellite intragénique codant. La taille normale est une répétition de la séquence CAG moins de 26 fois. Si le trinucléotide est répété entre 27 et 35 fois (allèle intermédiaire), la personne ne développera pas la maladie mais peut la transmettre à ses enfants. Si le trinucléotide est répété entre 36 et 40 fois (allèle mutant), la personne a de grands risques de développer la maladie. Si elle est supérieure à 40, la maladie surviendra assurément.
Il existe une corrélation entre le nombre de répétitions du codon et l'âge de survenue. Le nombre est supérieur à 60 dans la forme juvénile de Chorée de Huntington. L'homozygotie ne modifie pas l'âge de début de la maladie mais elle semble progresser plus rapidement. Le génotype ne parvient cependant pas à expliquer totalement l'âge d'apparition des premiers signes et il existe probablement des facteurs environnementaux qui ne sont pas connus[2].
La transmission se fait de manière autosomique dominante. Le phénomène d'anticipation apparaît surtout lors de la transmission paternelle. Ce phénomène d'anticipation apparaît au cours de la spermatogenèse. Les recoupements font penser que 3 couples anglais arrivés en 1630 en Nouvelle Angleterre seraient à l'origine de plus de 1000 cas de la maladie, répartis sur 12 générations[réf. nécessaire]. Ceci est l'exemple de l'effet fondateur d'une néomutation s'installant dans une population.
Ce gène IT15 code pour une protèine, la huntingtine dont le rôle n'est pas clair. Chez les patients atteints, la huntingtine est modifiée. Cette répétition est la seule mutation connue responsable de la Chorée de Huntington mais il semblerait que certains cas de chorée de Huntington aient été référencés ayant des IT15 dits normaux. Trois autres gènes sont suspectés: Prp, Junct-3 et TBP.
Anatomie pathologie
La dégénérescence neuronale est très tardive et lorsque les premiers signes apparaissent, elle débute depuis de nombreuses années (probablement dans l'enfance). Les lésions dans le cerveau sont caractérisées par une atrophie et une perte de neurones atteignant la région du striatum, essentiellement dans le noyau caudé et le putamen. D'autres régions cérébrales sont également concernées. Au microscope, il est observé à un stade précoce des inclusions dans le cytoplasme et les noyaux cellulaires contenant de la polyglutamine et de la huntingtine.
Incidence et prévalence
La prévalence de la maladie de Huntington varie selon les populations. Elle est estimée entre 3 et 7 pour 100 000 habitants en Europe de l'Ouest mais elle peut atteindre le nombre de 15 pour 100 000 habitants[3]. Cette prévalence est moindre en Chine, Japon, Finlande et dans les populations noires. Au Japon la prévalence est de 0,2 pour 100 000[réf. nécessaire], mais plus élevée dans la région du lac Maracaibo au Vénézuela où environ 700 personnes sont atteintes pour 100 000 habitants[1].
La disparité de prévalence entre les populations est expliquée car le nombre de répétitions du CAG (Cytosine-Adénine-Guanine) dans l'allèle de ces populations est moindre que les allèles des populations de l'Europe.
En France, 6 000 personnes sont atteintes de chorée de Huntington, soit environ 0.01% de la population, alors que 12 000 personnes seraient porteuses de la mutation mais asymptomatiques[1].
Description
La manière exacte dont la maladie affecte un individu est variable et peut même différer entre les membres d'une même famille, mais les symptômes progressent de façon prévisible pour la plupart des individus. Les premiers symptômes sont le manque général de coordination et une démarche instable. Lorsque la maladie progresse, des mouvements saccadés, non coordonnés du corps deviennent de plus en plus apparents, avec un déclin des aptitudes mentales et comportementales et des problèmes psychiatriques. Les aptitudes physiques sont progressivement diminuées jusqu'à ce qu'un mouvement coordonné devienne très difficile, et les capacités mentales évoluent généralement vers la démence. Bien que la maladie elle-même ne soit pas fatale, des complications comme la pneumonie, les maladies cardiaques, et des blessures dues à des chutes réduisent l'espérance de vie à environ vingt ans après l'apparition des symptômes. Il n'existe aucun remède contre la maladie et des soins à temps plein sont nécessaires dans les derniers stades de la maladie, mais il y a de nouveaux traitements pour soulager certains de ces symptômes.
L'âge moyen de début des signes est compris entre 40 et 50 ans. Le quart des malades verront les premiers signes apparaître après 50 ans, et quelques uns après 70 ans. Il existe une forme juvénile de chorée de Huntington qui débute avant 20 ans. Cette forme compte pour 10 % des malades atteints. Le nombre de répétition CAG est inversement proportionnel à l'âge d'apparition des premiers troubles moteurs : plus le nombre est grand et plus la maladie apparaît tôt.
Le début se manifeste par des troubles subtils de la coordination des mouvements (notamment oculaires) ou un changement d'humeur caractérisé par une tendance dépressive. Puis les mouvements anormaux deviennent évidents, entravant la vie professionnelle et obligeant souvent les personnes à abandonner leur travail. Des troubles cognitifs apparaissent, touchant plus l'organisation des tâches à effectuer que la mémoire. Un syndrome dépressif est fréquent pouvant conduire au suicide. Une dépendance s'installe. Tardivement les individus deviennent incontinents, muets et totalement dépendants pour la vie quotidienne. La mort survient en moyenne 15 à 25 ans après l'apparition des premiers signes. L'âge moyen du décès est de 55 ans.
Les mouvements anormaux (chorée), caractéristiques de la maladie, peuvent cependant être inconstants ou transitoires, diminuant lorsque le sujet devient atone. Ils ne sont donc pas corrélés avec le stade évolutif. Ils sont souvent absents dans les formes du sujet jeune, rendant le diagnostic plus difficile[3]. On notera que ces troubles moteurs sont majorés (et parfois mis en évidence) par les émotions et la concentration. L'incapacité de maintenir un tonus musculaire constant est par contre un marqueur de gravité. Elle se caractérise en particulier par une pression variable au cours d'une poignée de main[4].
Des convulsions peuvent survenir, surtout chez le sujet jeune.
Diagnostic
Le diagnostic est évoqué devant des troubles moteurs impliquant les mouvements volontaires et involontaires, des troubles cognitifs ou de l'humeur et une histoire familiale de maladie dominante. Ce point peut cependant manquer dans des proportions variables pouvant aller jusqu'à 25% des cas[5].
L'étude du cerveau par imagerie permet surtout d'éliminer les autres causes de démence. l'IRM et le scanner peuvent montrer des images non spécifiques et inconstantes : augmentation de la taille des cornes frontales des ventricules latéraux, atrophie du noyau caudé et du putamen constituant le striatum. Dans les stades tardifs, il se produit également une atrophie du cortex.
C'est une maladie à transmission autosomique dominante. Le diagnostic pouvant être établi avant même l'apparition des premiers symptômes, on parle de diagnostic présymptomatique[6].
Diagnostic différentiel
Le diagnostic différentiel se fait avec les différentes maladies se manifestant par une démence héréditaire, ou par des mouvements choréiques, soit :
- Démence frontotemporale
- Maladie de Huntington de type 2
- Formes génétiques de la maladie d'Alzheimer
- Maladie de Creutzfeldt-Jakob
- Neuroacanthocytose
Traitement
Si aucun traitement n'existe encore à ce jour, les espoirs de vaincre la maladie existent : recherche et expérimentation de traitements visant à améliorer voire restaurer les capacités de patients et recherche sur les greffes de neurones, embryonnaires notamment. La recherche sur la maladie de Parkinson peut aboutir à la découverte de traitements de la chorée.
Les organisations d'entraide, dont la première a été fondée dans les années 1960 augmentent en nombre. Elles travaillent pour accroître la sensibilité du public, pour fournir un soutien aux individus et à leurs familles et pour promouvoir la recherche. Ces organisations ont joué un rôle dans la recherche du gène en 1993.
Conseil génétique
Le conseil génétique est chargé d'informer et d'aider les personnes à envisager de passer les tests génétiques et est devenu un modèle pour d'autres maladies génétiquement dominantes.
Le test génétique n'est fait que pour une minorité des patients à risque[7], essentiellement en raison du refus de ces derniers à cause de l'absence de traitement et des conséquences psychologiques qui découlent du diagnostic. Le risque de suicide lors de l'annonce de ce dernier n'est d'ailleurs pas négligeable[8]. Le test génétique consiste à quantifier le nombre de répétition du gène IT15.
Famille d'un patient
Le dépistage pose des problèmes éthiques, notamment concernant l'âge à partir duquel un individu est considéré comme suffisamment mûr pour choisir de faire les tests, le droit des parents de faire tester leurs enfants et la confidentialité et la divulgation des résultats du test. La plupart des malades ont un parent atteint, les mutations de novo étant rares. Les pères asymptomatiques peuvent avoir un allèle intermédiaire ou avoir un allèle de pénétrance réduite (Séquence CAG entre 36-40). L'histoire familiale peut sembler négative en raison également de l'absence de diagnostic de la maladie, de décès survenue avant le début de la maladie ou de la survenue tardive de la maladie. L'intérêt du dépistage de la fratrie dépend du statut génétique des parents : si la répétition de la séquence CAG est supérieure à 41, le risque est de 1 sur 2. Si le père a un allèle intermédiaire le risque est de 5% (0,5 multiplié par 0,1). Un enfant qui hérite d'un allèle mutant à pénétrance réduite peut ou non développer la maladie
Pour les maladies héréditaires autosomiques dominantes, les enfants naissent toujours d'un parent porteur du même caractère. Le caractère apparaît à chaque génération en touchant autant de filles que de garçons. Un sujet atteint a la moitié de ses descendants atteints. Un sujet génétiquement sain donne une descendance saine. Seul le dépistage pré-implantatoire est possible à ce jour pour éviter de transmettre la maladie à ses enfants.
Média
Personnalités
Certaines personnalités telles que Sophie Daumier, Philippe Bedos (fils de Sophie Daumier) et Woody Guthrie (avant son fils Arlo Guthrie) sont atteintes de la chorée de Huntington.
Littérature et cinéma
- Le Test. Livre de Jean Baréma (éditions Lattès, 2002). Jean Baréma (nom d'emprunt) raconte dans cette autobiographie la trajectoire qui l'amène à faire le test de dépistage de la maladie.
- 10 ans avant ma mort, par Frédéric B. Récit autobiographique.
- Maudit gène, un documentaire de Anne Georget (2007). La réalisatrice suit le quotidien d'une famille touchée par la maladie et donne la parole à des médecins et chercheurs travaillant sur cette maladie. Liens associés : site du film (lien visité le 26 janvier 2008) ; fiche du film à l'IMDB.
- Dr. House, où l'un des personnages qui apparait à partir de la saison 4, le Dr Remy Hadley alias Numéro Treize, possède un parent atteint de la maladie et effectue le test en fin de saison. Celui-ci s'étant révélé positif, elle fait face à sa maladie dans la saison 5 en effectuant des tests cliniques avec le docteur Eric Foreman afin d'en déterminer l'évolution.
- Scrubs, où une des patientes du Dr Dorian (Zach Braff), dans le dernier épisode de la saison 8, est emmenée à l'hôpital par son voisin et montre des signes de démence visant son fils Dan, qui ne le reconnait plus. Dan décide quant à lui de ne pas faire les tests de dépistage pour la maladie lorsque qu'on lui annonce les symptômes et les évolutions de la maladie.
- Grey's Anatomy, saison 7 episode 4, une patiente dont la mère est morte de la maladie doit subir une intervention chirurgicale. Elle sait qu'elle a le gène de la maladie et est victime d'un premier symptôme: un tremblement incontrôlé de la main droite.
- Darkness, un film de Jaume Balaguero dans lequel le personnage principal est atteint du syndrome de Huntington
Notes et références
- Sandrine Humbert, « Maladie de Huntington : pourquoi les neurones meurent-ils ? », dans Pour la Science, no 383, septembre 2009 [résumé]
- (en)Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset Wexler NS, Lorimer J, Porter J et Als. Proc Natl Acad Sci U S A. 2004 Mar 9;101(10):3498-503
- (en)Huntington's disease, Francis O Walker, Lancet 2007; 369:218-228
- (en)Coordination of prehensile forces during precision grip in Huntington's disease, Gordon AM, Quinn L, Reilmann R, Marder K, Exp Neurol. 2000 May;163(1):136-48
- (en)High incidence rate and absent family histories in one quarter of patients newly diagnosed with Huntington disease in British Columbia, EW Almqvist, DS Elterman, PM MacLeod, MR Hayden, Clinical Genetics 2001;60(3);198–205
- Alexandra Dürr et Marcela Gargiulo, « Diagnostiquer une maladie avant qu'elle ne soit visible », dans Pour la science, no 383, septembre 2009 [résumé (page consultée le 30 août 2009)]
- (en)DNA analysis of Huntington’s disease : Five years of experience in Germany, Austria, and Switzerland, F. Laccone, U. Engel, E. Holinski–Feder, M. Weigell–Weber, K. Marczinek, D. Nolte, D. J. Morris–Rosendahl, C. Zühlke, K. Fuchs, H. Weirich–Schwaiger, G. Schlüter, G. von Beust, A. M. M. Vieira–Saecker, B. H. F. Weber, O. Riess, Neurology 1999;53:801
- (en)A Worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease, Elisabeth W. Almqvist, Maurice Bloch, Ryan Brinkman, David Craufurd, Michael R. Hayden,Am. J. Hum. Genet., 1999;64:1293-1304
- (en) Brendan Haigh, Mahbubul Huq, Michael R Hayden, Huntington Disease GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 [1]
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:143100 [2]
- (fr) Orphanet [PDF]
Liens externes
- Réseau Huntington de Langue Française Fédération d'équipes de recherche et de soin sur la maladie de Huntington.
- Association Huntington France Association francophone d'aide aux malades et aux familles
Wikimedia Foundation. 2010.