- Hamiltonien Moléculaire
-
Hamiltonien moléculaire
En optique, comme en chimie quantique, « hamiltonien moléculaire » est le nom donné au hamiltonien représentant l'énergie des électrons et des noyaux dans une molécule. Cet opérateur hermitien et l'équation de Schrödinger associée jouent un rôle central en chimie et en physique numériques pour calculer les propriétés des molécules et des agrégats de molécules, comme la conductivité, les propriétés optique et magnétique, ou encore la réactivité.
Introduction
On peut considérer que les briques d'une molécule sont les noyaux, caractérisés par leurs numéros atomiques Z, et les électrons de cœur, alors que les électrons dits « de valence », qui possèdent une charge élémentaire -q, en constituent le mortier (le liant). La charge d'un noyau de numéro atomique Z est Zq. Les électrons et les noyaux sont, dans une très bonne approximation, des charges et masses ponctuelles. Le hamiltonien moléculaire est une somme de nombreux termes : ses termes majeurs sont l'énergie cinétique des électrons et les interactions coulombiennes électrostatiques entre les deux types de particules chargées. Le hamiltonien qui contient seulement ces termes est appelé hamiltonien coulombien. Le hamiltonien moléculaire est complété avec des termes moins importants, la plupart d'entre eux étant imputables aux spins électroniques et nucléaires.
Bien qu'il soit généralement postulé que la solution de l'équation de Schrödinger dépendante du temps associée au hamiltonien de Coulomb prédise la plupart des propriétés d'une molécule, y compris sa forme (structure tridimensionnelle), les calculs basés sur un hamiltonien coulombien complet sont très rares, la raison principale étant que son équation de Schrödinger est très difficile à résoudre. Les applications sont donc restreintes aux petits systèmes comme la molécule de dihydrogène.
La grande majorité des calculs de fonctions d'ondes moléculaires sont basées sur la séparation du hamiltonien coulombien selon l'approximation de Born-Oppenheimer. Les termes d'énergie cinétique nucléaire[1] sont omis du hamiltonien coulombien et on considère le hamiltonien restant comme un hamiltonien purement électronique. Les noyaux stationnaires sont alors considérés dans le problème seulement comme générateurs d'un potentiel électrique dans lequel les électrons se meuvent selon les lois de la mécanique quantique. Dans ce cadre, le hamiltonien moléculaire a été simplifié en ce que l'on appelle le hamiltonien à noyau fixé ou encore hamiltonien électronique, qui agit selon des fonctions des coordonnées électroniques.
Une fois l'équation de Schrödinger du hamiltonien électronique résolue pour un nombre suffisant des « constellations » des noyaux, une valeur propre appropriée (en général la plus faible) peut être considérée comme fonction des coordonnées nucléaires, ce qui conduit de fait à une surface d'énergie potentielle. Dans les calculs pratiques, la surface est habituellement lissée en termes de fonctions analytiques. Dans la seconde étape de l'application de l'approximation de Born-Oppenheimer, la partie du hamiltonien coulombien complet qui dépend des électrons est remplacée par la surface d'énergie potentielle. Cela transforme le hamiltonien moléculaire en un autre opérateur hamiltonien qui agit seulement sur les coordonnées nucléaires. Dans le cas de la non-applicabilité de l'approximation de Born-Oppenheimer, ce qui se produite lorsque les énergies des différents états électroniques sont proches, les surfaces d'énergies potentielles avoisinantes sont nécessaires (se reporter à l'article pour plus de précisions).
L'équation de Schrödinger du mouvement nucléaire peut être résolue dans un référentiel fixe (le laboratoire), mais les énergies de translation et de rotation ne sont pas alors prises en compte. Seules les vibrations atomiques (internes) font partie du problème. De plus, pour des molécules plus importantes que des molécules triatomiques, il est relativement commun d'introduire l'approximation harmonique, qui approxime la surface d'énergie potentielle par une fonction quadratique des déplacements atomiques. Ceci donne le hamiltonien harmonique de mouvement nucléaire. Moyennant l'approximation harmonique, on peut convertir le hamiltonien en une somme de hamiltoniens monodimensionnels d'oscillateurs harmoniques. L'oscillateur harmonique monodimensionnel est l'un des rares systèmes permettant une résolution exacte de l'équation de Schrödinger.
L'équation de Schrödinger pour le mouvement nucléaire (rovibrationnel) peut être résolue de manière alternative dans un référentiel spécial, dit référentiel d'Eckart, qui translate et tourne avec la molécule. Formulé dans ce référentiel centré et fixé sur le corps étudié, le hamiltonien prend en compte la rotation, la translation et la vibration des noyaux. Depuis que Watson eut introduit une importante simplification de cet hamiltonien en 1968, on y réfère parfois sous le nom de hamiltonien de mouvement nucléaire de Watson, mais il est aussi connu sous le nom de hamiltonien d'Eckart.Hamiltonien coulombien
La forme algébrique de nombreuses observables (c'est-à-dire des opérateurs hermitiques représentant les quantités observables) est obtenue par les règles de quantification suivantes :
- écriture de la forme classique de l'observable dans la forme hamiltonienne (comme fonction du moment p et des positions q). Les deux vecteurs sont exprimés dans un référentiel galiléen, habituellement désignéc omme référentiel du laboratoire ou référentiel invariant dans l'espace.
- remplacement de p par
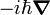 et interprétation de q comme un facteur multiplicatif. Ici
et interprétation de q comme un facteur multiplicatif. Ici  est l'opérateur nabla, opérateur vectoriel de dérivation au premier ordre. Les relations de commutations bien connues pour les opérateurs p et q découlent directement des règles de différentiation.
est l'opérateur nabla, opérateur vectoriel de dérivation au premier ordre. Les relations de commutations bien connues pour les opérateurs p et q découlent directement des règles de différentiation.
De manière classique, les électrons et les noyaux d'une molécule ont une énergie cinétique de la forme : p2/(2m) et interagissent par les forces de Coulomb, qui sont inversement proportionnelles à la distance rij entre la particule i et la particule j.
Dans cette expression ri désigne le vecteur de coordonnées de n'importe quelle particule (électron ou noyau). À partir d'ici, la lettre majuscule R représentera les coordonnées nucléaires, et la minuscule r les électrons du système. Les coordonnées peuvent être prises dans n'importe quel repère cartésien centré n'importe où dans l'espace, car la distance, comme produit interne, est invariante par rotation du référentiel et, comme norme d'un vecteur différentiel, également invariante par translation dudit référentiel.
En quantifiant l'énergie classique sous forme hamiltonienne on obtien un opérateur hamiltonien moléculaire parfois appelé hamiltonien coulombien. Ce hamiltonien est la somme de cinq classes de termes :
- les opérateurs d'énergie cinétique pour chaque noyau du système.
- les opérateurs dénergie cinétique pour chaque électron du système.
- les opérateurs d'énergie potentielle entre électrons et noyaux - attraction coulombienne totale dans le système.
- les opérateurs d'énergie potentielle de répulsions coulombiennes entre électrons.
- les opérateurs d'énergie potentielle de répulsions coulombiennes entre noyaux - aussi connue sous le nom d'énergie de répulsion nucléaire. Voir potentiel électrique.
Mi est ici la mass du noyau i, Zi le numéro atomique du noyau i, et me la masse de l'électron. L'opérateur laplacien de la particule i est :
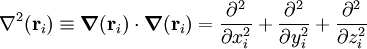 . L'opérateur d'énergie cinétique étant un produit interne, il est invariant par rotation dans un repère cartésien par rapport aux xi, yi, et zi exprimés. L'opérateur d'énergie cinétique, cependant, n'est pas invariant par translation (et donc du choix de l'origine du référentiel).
. L'opérateur d'énergie cinétique étant un produit interne, il est invariant par rotation dans un repère cartésien par rapport aux xi, yi, et zi exprimés. L'opérateur d'énergie cinétique, cependant, n'est pas invariant par translation (et donc du choix de l'origine du référentiel).Termes secondaires
Dans les années 1920 de nombreuses preuves spectroscopiques ont mis en évidence que le hamiltonien coulombien ne prenait pas en compte certains termes, qui bien que plus petits que les énergies cinétiques et coulombiennes, n'en sont pas moins non négligeables. Les observations spectroscopiques ont ainsi conduit à l'introduction d'un nouveau degré de liberté pour les électrons et noyaux, le spin. Ce concept empirique fut doté d'une base théorique par Paul Dirac lorsqu'il introduisit une forme relativiste correcte (invariance de Lorentz) dans l'équation de Schrödinger à une particule. L'équation de Dirac prédit que le spin et le mouvement spatial d'une particule interagissent via un couplage spin-orbite. Par analogie, le couplage spin-autre-orbite fut également introduit. Le fait qu'un spin de particule possède certaines caractéristiques d'un dipôle magnétique conduisit à proposer le couplage spin-spin. D'autres termes sans pendant classique sont : le terme de contact de Fermi (interaction de la densité électronique d'un noyau de taille finie avec un noyau) et le couplage quadrupolaire nucléaire (interaction d'un quadrupôle nucléaire avec le gradient d'un champ électrique du aux électrons).
Pour finir, un terme de violation de parité prédit par le modèle standard doit également être mentionné. Bien qu'il s'agisse d'une interaction extrêmement faible, elle a attiré l'attention des chercheurs comme le prouve la littérature scientifique car elle dote les énantiomères des molécules chirales d'énergies différentes.La suite de cet article ignorera les termes de spin et considèrera la solution de l'équation (de Schrödinger dépendante du temps) aux valeurs propres du hamiltonien coulombien.
Équation de Schrödinger liée au hamiltonien coulombien
Le hamiltonien coulombien a un spectre continu en raison du mouvement du centre de masse de la molécule dans un espace homogène. En mécanique classique, il est facile de distinguer le mouvement du centre de masse d'un système de masses ponctuelles. Son mouvement est classiquement découplé des autres mouvements. Il se déplace uniformément (avec une vitesse constante) dans l'espace comme s'il était une particule ponctuelle de masse égale à la somme Mtot des masses de toutes les particules.
En mécanique quantique, une particule libre possède comme fonction d'état une fonction d'onde plane, qui est une fonction qui n'est pas intégrable quadratiquement d'un moment clairement défini. L'énergie cinétique de cette particule appartient à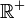 . La position du centre de masse est uniformément probable dans tout l'espace, selon le principe d'incertitude de Heisenberg.
. La position du centre de masse est uniformément probable dans tout l'espace, selon le principe d'incertitude de Heisenberg.
La séparation particulière du mouvement du centre de masse est, en mécanique quantique, beaucoup plus lourde qu'en mécanique classique. en introduicant le vecteur coordonnées du centre de masse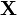 comme trois des degrés de liberté du système et en éliminant le vecteur coordonnée d'une particule (arbitraire), le nombre total de degrés de liberté restant donc le même, on peut obtenir par transformation linéaire un nouvel ensemble de coordonnées ti. Ces coordonnées sont des combinaisons linéaires des anciennes coordonnées de toutes les particules (noyaux et électrons). En appliquant le théorème de dérivation des fonctions composées, on peut montrer que :
comme trois des degrés de liberté du système et en éliminant le vecteur coordonnée d'une particule (arbitraire), le nombre total de degrés de liberté restant donc le même, on peut obtenir par transformation linéaire un nouvel ensemble de coordonnées ti. Ces coordonnées sont des combinaisons linéaires des anciennes coordonnées de toutes les particules (noyaux et électrons). En appliquant le théorème de dérivation des fonctions composées, on peut montrer que :Le premier terme de H est l'énergie cinétique du mouvement du centre de masse, qui peut être traité séparément, H' ne dépendant pas de
. Comme indiqué plus haut, ses états propres sont des ondes planes. Les constantes 1/μij sont positives et sont des combinaisons linéaires de toutes les masses inversées 1/mi. Elles constituent les masses réduites généralisées. Le potentiel 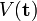 rassemble les termes de Coulomb exprimés dans les nouvelles coordonnées. Le premier terme de H' est similaire de manière habituelle à un opérateur énergie cinétique. Le second terme est connu sous l'appellation de terme de polarisation massique. Le hamiltonien invariant par translation H' peut être démontré comme étant auto-adjoint et lié à ce qui suit. Ceci étant dit, sa valeur propre la plus basse est réelle et finie. Bien que H' soit nécessairement invariant par permutations de particules identiques (car H et l'énergie cinétique du centre de masses sont invariants), son invariance n'est pas manifeste.
rassemble les termes de Coulomb exprimés dans les nouvelles coordonnées. Le premier terme de H' est similaire de manière habituelle à un opérateur énergie cinétique. Le second terme est connu sous l'appellation de terme de polarisation massique. Le hamiltonien invariant par translation H' peut être démontré comme étant auto-adjoint et lié à ce qui suit. Ceci étant dit, sa valeur propre la plus basse est réelle et finie. Bien que H' soit nécessairement invariant par permutations de particules identiques (car H et l'énergie cinétique du centre de masses sont invariants), son invariance n'est pas manifeste.
Peu d'applications moléculaires de H' existent actuellement. On pourra cependant se référer au travail fondamental sur la molécule d'hydrogène pour les premières applications[2]. Dans la grande majorité des calculs de fonctions d'ondes moléculaires le problème électronique est résolu grâce au hamiltonien à noyau fixé provenant de la première étape de l'approximation de Born-Oppenheimer.
On pourra se reporter à la référence[3] pour une discussion approfondie sur les propriétés mathématiques du hamiltonien coulombien. Dans ce papier, est aussi discuté ce qui peut arriver a priori au concept de molécule (comme système stable de noyaux et d'électrons avec une géométrie bien définie) à partir des propriétés du seul hamiltonien coulombien.Hamiltonien à noyau fixé
Le hamiltonien à noyau fixé décrit l'énergie des électrons dans le champ électrostatique des noyaux, où les noyaux sont postulés comme étant stationnaires par rapport à un référentiel galiléen.
La forme du hamiltonien électronique est :Les coordonnées des électrons et noyaux sont exprimés par rapport au référentiel qui se meut avec les noyaux, ce qui rend les noyaux immobiles par rapport à ce référentiel. Le référentiel reste parallèle à un référentiel fixé dans l'espace. Il s'agit d'un référentiel galiléen car les noyaux sont postulés comme non-accélérés par des forces ou couples externes. L'origine du référentiel est arbitraire, habituellement positionnée sur un noyau central où sur le centre de masse nucléaire. Parfois, il est postulé que les noyaux sont « au repos dans un référentiel fixe ». Ce postulat implique que les noyaux sont des particules classiques, car une particule quantique ne peut être au repos, ce qui signifierait qu'il aurait une quantité de mouvement nulle et une position définie, ce qui contredit le principe d'incertitude d'Heisenberg.
Les positions nucléaires étant constantes, l'opérateur énergie cinétique est invariant par translation selon tout vecteur nucléaire. Le potentiel coulombienne, dépendant des différences vectorielles, est aussi invariant. Dans la description des orbitales atomiques et pour le calcul des intégrales sur les orbitales atomiques, cette invariance est utilisée en dotant tous les atomes de la molécule avec leurs référentiels propres localisés parallèles au référentiel fixe.
Comme indiqué dans l'article approximation de Born-Oppenheimer, un nombre suffisant de solutions à l'équation de Schrödinger de Hel conduit à une surface d'énergie potentielle (SEP)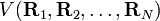 . Il est postulé que la dépendance fonctionnelle de V sur ses coordonnées est telle que :
. Il est postulé que la dépendance fonctionnelle de V sur ses coordonnées est telle que :pour
où t et s sont des vecteurs arbitraires et Δφ est un angle infinitésimal, Δφ >> Δφ2. Cette condition d'invariance sur la SEP est automatiquement remplie lorsque la SEP s'exprime en termes de différences de, et d'angles entre, les Ri,, ce qui est habituellement le cas.
Hamiltonien de mouvement nucléaire harmonique
Dans la suite de cet article, on considèrera la molécule comme semi-rigide. Dans la seconde étape de l'approximation de Born-Oppenheimer, l'énergie cinétique nucléaire Tn est réintroduite et l'équation de Schrödinger avec le hamiltonien :
est alors considérée. On pourra repérer dans sa solution : le mouvement du centre de masse nucléaire (3 degrés de liberté), la rotation globale de la molécule (3 degrés de liberté), et les vibrations nucléaires. En règle générale, ce n'est pas possible de le faire avec l'énergie cinétique nucléaire indiquée, cette dernière ne séparant pas explicitement les six degrés externes de liberté (rotation et translation globales) des 3N-6 degrés de liberté internes. En fait, l'opérateur d'énergie cinétique est ici défini par rapport à un référentiel fixe. Si l'on déplaçait l'origine du référentiel spatial fixe au centre de masse nucléaire, alors, par application de la loi de dérivation des fonctions composées, des termes de polarisation de masse nucléaire apparaitraient. Il est habituel d'ignorer ces termes dans leur globalité, ce qui sera fait par la suite.
Afin de parvenir à une séparation on devra distinguer les coordonnées internes et externes, ce pour quoi Eckart introduisit les conditions à remplir par les coordonnées. On montrera comment ces conditions apparaissent de manière naturelle à partir d'une analyse harmonique dans des coordonnées cartésiennes pondérées par masse.
Afin de simplifier l'expression pour l'énergie cinétique, on introduit les coordonnées pondérées de déplacement :
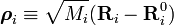 .
.
Puisque :
l'opérateur d'énergie cinétique devient :
Si l'on effectue un développement de Taylor de V au voisinage de la géométrie d'équilibre,
et que l'on effectue une troncature après trois termes (c'est-à-dire que l'on effectue une approximation harmonique), on peut décrire V avec seulement le troisième terme. Le terme V0 peut être absorbé dans l'énergie (donnant un nouveau zéro). Le deuxième terme disparaît en raison de la condition d'équilibre. Le terme restant contient la matrice hessienne F de V, qui est symétrique et peut être diagonalisée avec une matrice orthogonale 3N x 3N avec des éléments constants :
Il peut être montré à partir de l'invariance de V par rotation et translation que six des vecteurs propres de F (les six dernières lignes de Q) ont pour valeur propre 0 (sont des modes de fréquence nulle). Ils décrivent l'espace extérieur.
Les premières 3N-6 lignes de Q sont - pour des molécules dans leurs états fondamentaux - des vecteurs propres à valeurs propres non nulles ; elles sont les coordonnées internes et forment une base orthonormée pour un sous-espace de dimension 3N - 6 de l'espace des configurations nucléaires R3N, l'espace interne. Les vecteurs propres de fréquence nulle sont orthogonaux aux vecteurs propres de fréquences non nulles. Il peut être démontré que ces orthogonalités correspondent en fait aux conditions d'Eckart. L'énergie cinétique exprimée dans les coordonnées internes est l'énergie cinétique interne (vibrationnelle).Avec l'introduction des coordonnées normales :
la partie vibrationnelle (interne) du hamiltonien pour le mouvement nucléaire devient dans l'approximation harmonique
L'équation de Schrödinger correspondante est aisément résolue, en étant factorisée en 3N-6 équations pour des oscillateurs harmoniques mono dimensionnels. La difficulté principale dans cette approximation de la solution de l'équation de Schrödinger du mouvement nucléaire est le calcul du hessien F de V et sa diagonalisation.
Cette approximation du problème du mouvement nucléaire, décrit dans les 3N coordonnées pondérées cartésiennes, est devenu standard en chimie quantique depuis (années 1980-1990) que les algorithmes pour des calculs précis du hessien F sont disponibles. En dehors de l'approximation harmonique, il est problématique que les mouvements externes (rotation et translation) de la molécule ne soient pas considérés. Ils sont pris en compte dans un hamiltonien rovibrationnel qui est parfois appelé hamiltonien de Watson.Hamiltonien du mouvement nucléaire de Watson
Afin d'obtenir un hamiltonien pour les mouvements externes (translation et rotation) couplés aux mouvements internes (vibration), il est usuel de faire un retour à ce niveau à la mécanique classique et de formuler lénergie cinétique classique correspondant à ces mouvements des noyaux. Il est facile de séparer de manière classique le mouvement de translation (du centre de masse) des autres mouvements. Cependant, séparer le mouvement de rotation du mouvement vibrationnel est plus difficile, et en réalité, pas entièrement possible. Cette séparation rovibrationelle fut pour la première fois réalisée par Eckart[4] en 1935 en imposant ce qui est maintenant connu sous le nom de conditions d'Eckart. Lorsque le problème est décrit dans un référentiel (dit d'Eckart) qui tourne avec la molécule, et donc non galiléen, les énergies associées aux forces d'inertie (force centrifuge et force de Coriolis) apparaissent dans l'énergie cinétique.
En général, l'énergie cinétique classique T définit le tenseur métrique g = (gij) associé avec les coordonnées curvilignes s = (si) par la relation :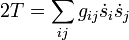 .
.
L'étape de quantification est la transformation de cette énergie cintéique classique en un opérateur de chimie quantique. Il est usuel de suivre la méthode proposée par Podolsky[5] en réécrivant l'opérateur de Laplace-Beltrami dans les mêmes coordonnées s (généralisées, curvilignes) utilisées pour la forme classique. L'équation pour cet opérateur nécessite l'inversion du tenseur métrique g et son déterminant. La multiplication de l'opérateur de Laplace-Beltrami par
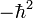 procure l'opérateur d'énergie cinétique quantique requis. Lorsque l'on applique cette méthode aux coordonnées cartésiennes, qui ont une métrique unitaire, la même énergie cinétique est obtenue par application des règles de quantification.
procure l'opérateur d'énergie cinétique quantique requis. Lorsque l'on applique cette méthode aux coordonnées cartésiennes, qui ont une métrique unitaire, la même énergie cinétique est obtenue par application des règles de quantification.
Le hamiltonien de mouvement nucléaire a été décrit par Wilson et Howar en 1936[6], qui suivirent cette méthode, puis il fut redéfini plus tard par Darling et Dennison en 1940[7]. Cette méthode resta le standard jursqu'en 1968, lorsque Watson[8] fut capable de le simpflier de manière drastique en commutant par les dérivées le déterminant du tenseur métrique. On donnera par la suite le hamiltonien rovibrationnel obtenu par Watson, parfois appelé hamiltonien de Watson. On se doit de préciser que la dérivation de ce Hamiltonien est aussi possible en commençant depuis l'opérateur laplacien dans sa forme cartésienne, en appliquant les transformations de coordonnées, puis en utilisant les règles des dérivation des fonctions composées[9]. Le hamiltonien de Watson, décrivant tous les mouvements des N noyaux, est :Le premier terme est celui du centre de masse
Le second terme est le terme rotationnel semblable à l'énergie cinétique du rotateur rigide. Ici,
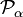 est la composante α de l' opérateur de moment angulaire du rotateur rigide fixe[10]. L'opérateur
est la composante α de l' opérateur de moment angulaire du rotateur rigide fixe[10]. L'opérateur 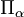 est une composante de l'opérateur dconnu sous le nom d'opérateur de moment angulaire vibrationnel (bien qu'il ne satisfasse pas aux relations de commutation du moment angulaire),
est une composante de l'opérateur dconnu sous le nom d'opérateur de moment angulaire vibrationnel (bien qu'il ne satisfasse pas aux relations de commutation du moment angulaire),avec la constante de couplage de Coriolis :
Ici, εαβγ est le symbole de Levi-Civita. Les termes quadratiques dans
sont des termes centrifuges, les termes bilinéaires dans et 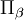 sont des termes de Coriolis.
sont des termes de Coriolis.
Les quantités Q s, iγ sont des composantes des coordonnées normales introduites ci-dessus. Les coordonnées normales peuvent être introduites de manière alternative par l'application de la méthode GF de Wilson. La matrice symétrique 3 x 3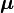 est appelée tenseur effectif réciproque d'inertie. Si tous les qs étaient nuls (molécule rigide), le référentiel d'Eckart coinciderait avec le repère des axes principaux (voir rotateur rigide) et serait diagonale, avec les moments d'inertie réciproques d'équilibre sur la diagonale. Si tous les qs étaient nuls, seule les énergies cinétiques de translation et de rotation rigide seraient exprimées.
est appelée tenseur effectif réciproque d'inertie. Si tous les qs étaient nuls (molécule rigide), le référentiel d'Eckart coinciderait avec le repère des axes principaux (voir rotateur rigide) et serait diagonale, avec les moments d'inertie réciproques d'équilibre sur la diagonale. Si tous les qs étaient nuls, seule les énergies cinétiques de translation et de rotation rigide seraient exprimées.
Le terme U de type potentiel est le terme de Watson :proportionnel à la trace du tenseur effectif réciproque d'inertie. Le quatrième terme dans le hamiltonien de Watson est l'énergie cinétique associée aux vibrations atomiques (nucléaires) exprimée en coordonnées normales qs, qui comme indiqué ci-dessus, sont données en termes de déplacements nucléaires ρiα par :
Enfin, V est l'énergie potentiel non développée par définition dépendante des seules coordonnées internes. Dans l'approximation harmonique, elle prend la forme :
Notes et références
- ↑ Parfois qualifiée de « ionique », mais cette appellation est plus présente en physique du solide.
- ↑ W. Kołos and L. Wolniewicz, Nonadiabatic Theory for Diatomic Molecules and Its Application to the Hydrogen Molecule, Rev. Mod. Phys. vol. 35, pp. 473-483 (1963)
- ↑ R. G. Woolley and B. T. Sutcliffe, P.-O. Löwdin and the Quantum Mechanics of Molecules in Fundamental World of Quantum Chemistry, E. J. Brändas and E. S. Kryachko (Eds.), Vol. 1, 21-65, Kluwer Academic Publshers, 2003.
- ↑ C. Eckart, Some studies concerning rotating axes and polyatomic molecules, Physical Review, vol. 47, pp. 552-558 (1935).
- ↑ B. Podolsky, Quantum-mechanically correct form of Hamiltonian function for conservative system, Phys. Rev., vol. 32, p. 812 (1928)
- ↑ E. Bright Wilson, Jr. and J. B. Howard The Vibration-Rotation Energy Levels of Polyatomic Molecules I. Mathematical Theory of Semirigid Asymmetrical Top Molecules, J. Chem. Phys. vol. 4, pp. 260-268 (1936)
- ↑ B. T. Darling and D. M. Dennison, The water vapor molecule, Phys. Rev., vol. 57, pp. 128-139 (1940).
- ↑ J. K. G. Watson, Simplification of the molecular vibration-rotation Hamiltonian, Mol. Phys. vol. 15, 479-490 (1968)
- ↑ L. C. Biedenharn and J. D. Louck, Angular Momentum in Quantum Physics, volume 8 of Encyclopedia of Mathematics, Addison-Wesley, Reading, 1981.
- ↑ On pourra se référer à l'article Matrice D de Wigner pour avoir son expression en termes d'angles d'Euler.
Pour plus de renseignements
- Born, M. et Oppenheimer, R., « Zur Quantentheorie der Molekeln », dans Annalen der Physik, vol. 84, 25 août 1927, p. 457-484
- Moss, R.E., Advanced Molecular Quantum Mechanics, Chapman and Hall
- Tinkham, M., Group Theory and Quantum Mechanics, Dover Publications (ISBN 0486432475)
- Discussion sur les termes de spin dans le hamiltonien moléculaire : R. McWeeny, Methods of Molecular Quantum Mechanics, 2nd edition, Academic, London (1989).
Voir aussi
- Approximation de Born-Oppenheimer
- Conditions d'Eckart
- Méthode GF
- Principe Franck-Condon
- Rotateur rigide
- Transformation adiabatique (chimie quantique)
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « Molecular Hamiltonian ».
 Portail de la chimie
Portail de la chimie Portail de la physique
Portail de la physique
Catégories : Chimie quantique | Spectroscopie
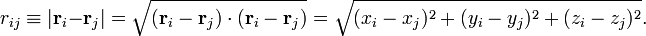
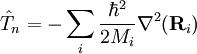
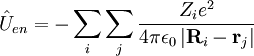
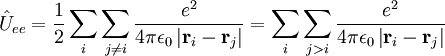
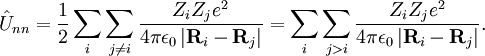
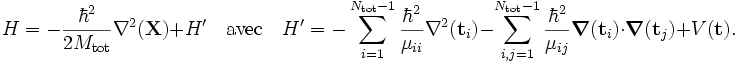
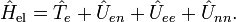
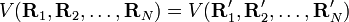
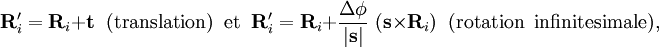
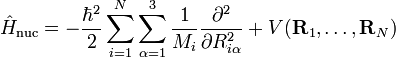
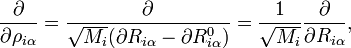
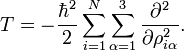
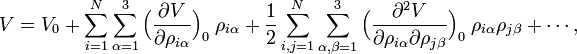
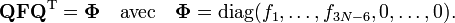
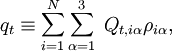
![\hat{H}_\mathrm{nuc} \approx \frac{1}{2} \sum_{t=1}^{3N-6} \left[-\hbar^2 \frac{\partial^2}{\partial q_{t}^2} + f_t q_t^2 \right] .](/pictures/frwiki/51/30d9b5a62d763b4596dbd352549bef55.png)
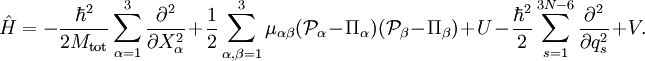
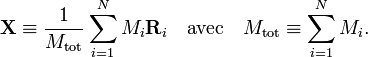
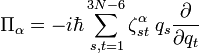
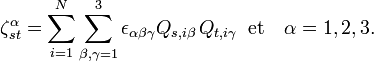
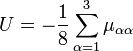
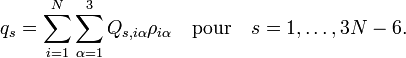
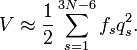
Wikimedia Foundation. 2010.