- Polythiophène
-
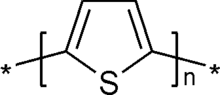 Unité de répétition monomère de polythiophène non substitué. Les étoiles indiquent les groupes de terminaison de la chaîne polymère.
Unité de répétition monomère de polythiophène non substitué. Les étoiles indiquent les groupes de terminaison de la chaîne polymère.
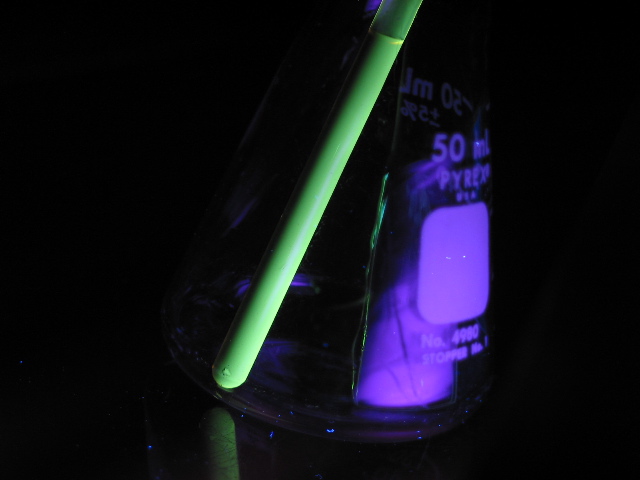
 Les polythiophènes montrent des propriétés optiques résultant de leur structure conjuguée, comme le démontre la fluorescence d'une solution de polythiophène substitué sous irradiation UV.
Les polythiophènes montrent des propriétés optiques résultant de leur structure conjuguée, comme le démontre la fluorescence d'une solution de polythiophène substitué sous irradiation UV.Les polythiophènes (PT) constituent une famille de polymères (macromolécules) résultant de la réaction de polymérisation du thiophène, un hétérocycle sulfuré, qui peut devenir conducteur lorsque des électrons sont ajoutés ou enlevés des orbitales p conjuguées par dopage.
La propriété la plus remarquable de ces matériaux, la conductivité électrique, est une résultante de la délocalisation électronique le long de la chaîne polymère - d'où parfois leur qualification de « métaux synthétiques ». Cependant, elle ne constitue pas la seule propriété intéressante due à cette délocalisation des électrons. Les propriétés optiques dépendent en effet des stimuli environnementaux, avec des modifications drastiques de couleur selon le solvant, la température, le potentiel appliqué, et les liaisons à d'autres molécules. Les changements de couleurs et de conductivité sont induits par le même mécanisme - la torsion du squelette polymère, rompant la conjugaison - ce qui fait d'eux des capteurs chimiques donnant une large gamme de réponses électroniques et optiques.
Sommaire
Bref historique
L'étude des polythiophènes - et plus largement sur les polymères conducteurs - s'est essentiellement déroulée sur les trois dernières décennies jusqu'à voir son excellence confirmée par l'obtention en 2000 du prix Nobel de chimie par Alan Heeger, Alan MacDiarmid et Hideki Shirakawa « pour la découverte et le développement de polymères conducteurs ». Un nombre important d'articles de revue a déjà été publié sur les PT, le premier d'entre eux datant de 1981[1]. Schopf et Koßmehl ont ainsi publié une revue claire de la littérature publiée entre 1990 et 1994[2]. Roncali observa la synthèse par voie électrochimique en 1992[3], et les propriétés électroniques des PT substitués en 1997[4]. La revue de McCullough de 1998 s'appesantissait sur les synthèses chimiques de PT conducteurs[5]. Une revue générale sur les polymères conjugués des années 1990 a été menée par Reddinger et Reynolds en 1999[6]. Enfin, Swager et al. ont étudié des capteurs chimiques basés sur des polymères conjugués en 2000[7]. Ces articles de revue sont d'excellents guides pour mettre en lumière les premières publications sur les PT des deux dernières décennies.
Mécanisme de conduction et de dopage
Les électrons sont délocalisés le long des chaînes conjuguées des polymères conducteurs, ordinairement par la superposition d'orbitales p, ce qui aboutit à un système π étendu avec une bande de valence pleine. En ôtant des électrons du système π (dopage p), ou en ajoutant (dopage n), une unité chargée appelée bipolaron est formée (voir figure 1).
 Figure 1. Départ de deux électrons d'une chaîne PT (dopage p) formant un bipolaron.
Figure 1. Départ de deux électrons d'une chaîne PT (dopage p) formant un bipolaron.Le dopage est produit à des taux importants (de 20 à 40 %) dans les polymères conducteurs que dans les semi-conducteurs (< 1%). Le bipolaron bouge comme une entité unique le long de la chaîne polymère, et est responsable de la conductivité du polymère qui peut être observée macroscopiquement. Sur certains échantillons de poly(3-dodécylthiophène) dopé à l'iode, la conductivité peut atteindre 1000 S/cm[8]. (en comparaison, la conductivité du cuivre est approximativement de 5.105 S/cm). Généralement, la conductivité des PT est inférieure à 1000 S/cm, mais une conductivité plus élevée n'est pas nécessaire pour de nombreuses applications de polymères conducteurs (voir ci-dessous pour des exemples).
L'oxydation du polymère conducteur et l'introduction de contre-ions (dopage p) simultanément, peuvent être accomplis par voie chimique ou électrochimique. Durant la synthèse électrochimique d'un PT, les contre-ions dissous dans le solvant peuvent s'associer avec le polymère lorsqu'il est déposé sur l'électrode dans sa forme oxydée. En dopant le polymère lors de sa synthèse, une couche mince peut être déposée sur l'électrode - le polymère conduit les électrons du substrat vers la surface du film. De manière alternative, un film ou une solution de polymère conducteur neutre peut être dopé en post-synthèse.
La réduction du polymère conducteur (dopage n), est beaucoup moins courante que le dopage p. Une étude préliminaire du dopage électrochimique a montré que les niveaux de dopage n étant moins nombreux que ceux de dopage p, les cycles de dopage n sont moins efficaces, le nombre de cycles requis pour atteindre le dopage maximal est plus élevé, et le processus de dopage n apparaît comme être cinétiquement limité, en raison de la diffusion des contre-ions dans le polymère[9].
Une large gamme de réactifs ont été utilisés pour doper les PT. L'iode et le brome produisent des conductivités élevées[8] mais sont instables et s'évaporent doucement du matériau[10]. Les acides organiques, comme l'acide trifluoroacétique, l'acide propanoïque et des acides sulfoniques produisent des PT avec des conductivités inférieures à celles produites par les halogènes, mais avec des stabilités environnementales supérieures[10],[11]. La polymérisation oxydante avec du chlorure ferrique peut entraîner un dopage par le catalyseur résiduel[12] bien que des études en spectrométrie de masse MALDI aient montré que les poly(3-hexylthiophène)s sont aussi partiellement halogénés par l'agent oxydant résiduel[13]. Le poly(3-octylthiophène) dissous dans du toluène peut être dopé par des solutions d'hexahydrate de chlorure ferrique dissous dans de l'acétonitrile, et peut être déposé en films avec des conductivités atteignant 1 S/cm[14]. On peut citer parmi d'autres dopants p moins communs le chlorure d'or(III)[15] et l'acide trifluorométhanesulfonique[16].
Propriétés structurales et optiques
Longueur de conjugaison et chromismes
Les systèmes p étendus de PT conjugués produisent une des propriétés les plus intéressantes de ces matériaux, leurs propriétés optiques. En première approximation, le squelette conjugué peut être considéré comme un exemple réel de la solution de l'« électron dans une boîte » de l'équation de Schrödinger; cependant, le développement de modèles plus fins pour prédire de manière fine les spectres d'absorption et de fluorescence de systèmes d'oligo-thiophènes bien définis est en cours[17]. La conjugaison repose sur la superposition des orbitales p des cycles aromatiques, qui, en retour, nécessite que les cycles de thiophène soient coplanaires (voir figure 2, en haut).
 Figure 2. Orbitales p conjuguées d'un PT coplanaire et substitué.
Figure 2. Orbitales p conjuguées d'un PT coplanaire et substitué.Le nombre de cycles coplanaires détermine la longueur de conjugaison : plus grande est la longueur de conjugaison, plus la séparation entre niveaux d'énergie sera faible et plus la longueur d'onde d'absorption sera importante. La déviation vis-à-vis de la coplanarité peut être permanente, résultant de mauvaises liaisons durant la synthèse ou être provoquées par l'adjonction de chaînes latérales volumineuses (gêne stérique), ou temporaire, résultant de modifications de l'environnement ou des liaisons. Cette torsion du squelette réduit la longueur de conjugaison (voir figure 2, en bas) et la séparation des niveaux d'énergie s'accroît, ce qui réduit la longueur d'onde d'absorption.
La détermination de la longueur de conjugaison maximale effective nécessite la synthèse de PT réguliers par régions de longueur définie. La bande d'absorption dans la région du visible est décalée vers le rouge lorsque la longueur de conjugaison augmente, et la longueur de conjugaison maximale effective est calculée comme le point de saturation de décalage vers le rouge. Les premières études de Ten Hoeve et al. ont indiqué que la conjugaison effective s'étend sur 11 unités de répétition[18], puis d'autres études ultérieures ont accru cette estimation à 20 unités[19]. Plus récemment, Otsubo et al. ont effectué les synthèses d'oligothiophènes à 48[20] et 96[21], et ont découvert que le décalage dans le rouge, tant qu'il est faible (une différence de 0,1 nm entre le 72- et le 96-mère), ne sature pas, ce qui signifie que la longueur de conjugaison effective peut être plus importante même que 96 unités[21].
Une large gamme de facteurs environnementaux peuvent causer la torsion du squelette conjugué, réduisant ainsi la longueur de conjugaison et provoquant le décalage de la bande d'absorption, incluant les solvants, la température, l'application d'un champ électrique ou les ions dissous. La bande d'absorption du poly(3-thiophène acide acétique) dans une solution aqueuse d'alcool polyvinylique (PVA) est décalée de 480 nm à pH 7 à 415 nm à pH 4. Cela peut être attribué à la formation d'une structure de pelote compacte pouvant former des liaisons hydrogène avec le PVA lors d'une déprotonation partielle du groupe acide acétique[22]. Les PT chiraux ne montre pas de dichroïsme circulaire induit (ICD) dans le chloroforme, mais démontre des ICD intenses mais opposés dans des mélanges chloroforme-acétonitrile et chloroforme-acétone[23]. Ainsi, un PT avec un acide aminé chiral comme chaîne latérale[24] montre des décalages de bande d'absorption et des ICD modérés en fonction du pH et de la concentration du tampon[25].
Les décalages dans les bandes d'absorption des PT en raison d'une modification de température résultent d'une transition conformationnelle d'une conformation coplanaire quasi-linéaire à basse température à une conformation non-plane, en pelote pour des températures plus élevées. Par exemple, le poly(3-(octyloxy)-4-méthylthiophène) subit un changement de couleur du rouge violacé à 25 °C au jaune pâle à 150 °C. Un point isobestique (point auquel les courbes d'absorption à toutes les températures se superposent) indique la coexistence entre deux phases, qui peuvent exister pour une même chaîne ou pour différentes chaînes[26]. Cependant, tous les PT thermochromes ne présentent pas de point isobestique : les très régioréguliers (la régiorégularité est le fait que les monomères s'agencent de la même manière sur la plus grande longueur de chaîne possible et le plus fréquemment possible) poly(3-alkylthiophène)s (PAT) montre un décalage vers le bleu continu lorsque la température augmente si les chaînes sont assez courtes pour ne pas se mêler et s'interconvertir entre phases cristallines et désordonnées à basse température[réf. nécessaire].
Enfin, les PT peuvent montrer des décalages d'absorption dus à l'application de champs électriques (électrochromisme)[27] ou à l'introduction d'ions alcalins (ionochromisme)[28]. Ces effets seront discutés ci-dessous dans le contexte de l'application des PT.
Régiorégularité
L'asymétrie des thiophènes trisubstitués conduit à trois couplages possibles lorsque deux monomères sont reliés entre les positions 2 et 5. Ces couplages sont :
- 2,5' ou couplage tête-queue (en anglais head–tail - HT)
- 2,2' ou couplage tête-tête (en anglais head–head - HH)
- 5,5' ou couplage queue-queue (en anglais tail–tail - TT)
Ces trois diades peuvent se combiner en quatre triades distinctes, indiquées figure 3.
 Figure 3.Les quatre triades possibles résultant du couplage de thiophènes triplement substitués.
Figure 3.Les quatre triades possibles résultant du couplage de thiophènes triplement substitués.Les triades peuvent être distinguées par spectroscopie RMN, et le degré de régiorégularité peut être estimé par intégration du spectre obtenu[29],[30].
Elsenbaumer et al. remarquèrent en premiers l'effet de la régiorégularité sur les propriétés des PT : un copolymère de 3-méthylthiophène et de 3-butylthiophène à blocs aléatoires possède ainsi une conductivité de 50 S/cm, alors qu'un copolymère plus régiorégulier avec un rapport de 2/1 de couplages HT et HH possède une conductivité plus importante de 140 S/cm [31]. Des films de poly(3-(4-octylphényl)thiophène) (POPT) régiorégulier avec plus de 94 % de HT possèdent des conductivités de 4 S/cm, à comparer avec les 0,4 S/cm du POPT régioirrégulier[32]. Les PAT préparés en utilisant du zinc de Rieke forment « des films cristallins, flexibles, et couleur bronze avec un éclat métallique ». D'un autre côté, les polymères régioaléatoires correspondants produisent des « films amorphes et orange »[33]. La comparaison des propriétés thermochromes des PAT de Rieke montre que, alors que les polymères régioréguliers montrent des effets thermochromes forts, les régioirréguliers ne présentent pas de changements significatifs à température élevée. Ceci est probablement dû à la formation de défauts conformationnels légers et localisés[réf. nécessaire]. Enfin, Xu et Holdcroft ont montré que l'absorption de fluorescence et les maxima d'absorption des poly(3-hexylthiophène)s se produisent à des longueurs d'ondes décroissantes (plus haute énergie) lorsque le taux de diades HH croît. La différence entre l'absorption et les maxima d'absorption, le déplacement de Stokes, croît aussi avec le taux de diades HH, ce qui peut être attribué principalement à la contrainte conformationnelle dans le premier état excité[34].
Solubilité
Les PT non substitués sont conducteurs après dopage, et possèdent une excellente stabilité experimentale, comparés à d'autres polymères conducteurs comme le polyacétylène (ou polyéthyne selon la nomenclature IUPAC : (C2H2)n), mais sont peu manipulables et solubles uniquement dans des solutions comme les mélanges de trifluorure et de pentafluorure d'arsenic[35]. Cependant, en 1987, des exemples de PT organo-solubles ont été annoncés. Elsenbaumer et al., en utilisant un couplage croisé de Grignard catalysé par du nickel, ont pu synthétiser deux PT solubles, poly(3-butylthiophène) et poly(3-méthylthiophène-'co'-3’-octylthiophène), qui peuvent être déposés en films et dopés à l'iode pour atteindre des conductivités de 4 à 6 s/cm[36]. Hotta et al. ont synthétisé par voie électrochimique du poly(3-butylthiophène) et du poly(3-hexylthiophène)[37] (et posterieurement par voie chimique[38]) et caractérisé ces polymères en solution[39], puis déposé en films[40]. Les PAT solubles montrent à la fois du thermochromisme et du solvachromisme (voir ci-dessus) dans le chloroforme et le 2,5-diméthyltétrahydrofurane[41].
Également en 1987, Wudl et al. ont réussi les synthèses de poly(3-thiophènealcanesulfonate)s hydrosolubles[42]. En plus de leur conférer l'hydrosolubilité, les groupes latéraux sulfonates agissent comme des contre-ions, et induisent donc un auto-dopage de ces polymères conducteurs. Les PT substitués avec des fonctions acides carboxyliques[43], acides acétiques[44], acides aminés[24], et uréthanes[45] sont aussi hydrosolubles.
Plus récemment, des poly(3-(perfluorooctyl)thiophène)s solubles dans du dioxyde de carbone supercritique[46] ont été synthétisés par voies chimie et électrochimique par Collard et al.[47]. Enfin, des oligothiophènes non substitués terminés à chaque extrémité par des esters d'alkyles thermiquement labiles ont été déposés en film à partir de solution, puis chauffés afin d'ôter leurs groupes terminaux solubilisants. Des images de microscopie à force atomique (AFM) ont montré un accroissement significatif de l'ordre à longue portée après chauffage[48].
Synthèse
Les PT peuvent être synthétisé par voie électrochimique, en leur appliquant un potentiel sur une solution de monomères afin de provoquer la polymérisation, ou chimiquement, en utilisant des oxydants ou des catalyseurs de couplage croisé. Ces deux méthodes présentent leurs avantages et désavantages.
Synthèse électrochimique
Lors d'une polymérisation électrochimique, un potentiel électrique est appliqué à une solution contenant du thiophène et un électrolyte, produisant ainsi un film PT conducteur à l'anode[réf. nécessaire]. La polymérisation électrochimique est intéressante, le polymère n'ayant pas besoin d'être isolé puis purifié, mais elle produit des structures avec des degrés variables d'irrégularités structurales, comme de la réticulation.
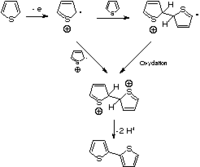 Figure 4.Premières étapes de la polymérisation du thiophène.
Figure 4.Premières étapes de la polymérisation du thiophène.
Comme le montre la figure 4, l'oxydation d'un monomère produit un radical cation, qui peut être apparié avec un second radical cation pour former un dimère dication, ou avec un autre monomère pour produire un dimère cationique radicalaire. De nombreuses techniques, comme la microscopie vidéo in situ[49], la voltampérométrie cyclique[50], la spectroscopie de photocourant[51], ou des mesures électrochimiques par microbalance à quartz[52], ont été utilisées afin de comprendre les mécanismes de nucléation et de croissance conduisant au dépôt du polymère sur l'anode. Le dépôt de longues chaînes bien ordonnées sur la surface de l'électrode est suivie de la croissance de chaînes soit longues et flexibles soit plus courtes et plus réticulées, selon les conditions de polymérisation.La qualité d'un film de PT préparé par voie électrochimique est dépendante de nombreux facteurs. Cela inclut la matériau d'électrode, la densité, la température, le solvant, l'électrolyte, la présence d'eau et la concentration en monomère[2]. Deux autres facteurs importants mais interagissants sont la structure du monomère et le potentiel appliqué. Le potentiel requis pour l'oxydation du monomère dépend de la densité électronique dans le système p du cycle du thiophène. Les groupes donneurs d'électrons abaissent le potentiel d'oxydation, les groupes accepteurs l'augmentant. En conséquence, le 3-méthylthiophène polymérise dans l'acétonitrile et le tétrafluoroborate de tétrabutylammonium à un potentiel d'environ 1,5 V avec une électrode au calomel saturé (ECS), tandis que le thiophène non substitué polymérise à environ 1,7 V avec une ECS.
La gêne stérique résultant de l'adjonction sur le carbone a d'un thiophène tri-substitué inhibe la polymérisation[53]. Cette observation conduit au phénomène appelé « paradoxe polythiophène » : le potentiel d'oxydation de nombreux monomères de thiophène est plus élevé que le potentiel d'oxydation du polymère résultant. En d'autres termes, le polymère peut être oxydé de manière irréversible et se décompose à un taux comparable à la polymérisation du monomère correspondant[réf. nécessaire]. Cela reste un des défauts majeurs de la polymérisation électrochimique, et limite son application pour de nombreux monomères thiophéniques avec des groupes latéraux complexes.
Synthèse chimique
La synthèse chimique des PT présente deux avantages comparé à la voie électrochimique : une meilleure sélection des monomères, et, en utilisant des catalyseurs appropriés, la possibilité de produire des PT substitués parfaitement régioréguliers. Lorsque les PT pourraient avoir été synthétisés chimiquement par accident il y a plus d'un siècle[54], les premières synthèses planifiées utilisant une polymérisation métal-catalysée du 2,5-dibromothiophène ont été reportées de manière indépendante par deux groupes en 1980. Yamamoto et al. ont utilisé du magnésium dans le tétrahydrofurane (THF) et du nickel-dichlorure de bipyridine, analogue au couplage de Kumada des réactifs de Grignard pour les halogénures d'aryles[55]. Lin et Dudek ont également utilisé du magnésium dans le THF, mais avec une série de catalyseurs acétylacétonate (Pd(acac)2, Ni(acac)2, Co(acac)2, et Fe(acac)3)[56].
Des développements ultérieurs ont produit des PT de plus grande masse molaire que ceux des travaux initiaux, et peuvent être groupés en deux catégories en se basant sur leurs structures. Les PT régioréguliers peuvent être synthétisés par des réactions de couplages croisés catalysés de bromothiophènes, alors que les polymères avec divers degrés de régiosélectivité peuvent être simplement synthétisés par une polymérisation oxydante.
 Figure 5. Méthode de couplages croisés pour la préparation des PAT.
Figure 5. Méthode de couplages croisés pour la préparation des PAT.La première synthèse de PAT régioréguliers fut décrite par McCullough et al. in 1992[57]. Comme le montre la figure 5 (en haut), une bromation sélective produit du 2-bromo-3-alkylthiophène, suivie par une transmétallation puis un couplage croisé de Kumada en présence de nickel comme catalyseur. Cette méthode produit environ 100 % de couplages HT–HT, selon l'analyse spectroscopique RMN des diades. Dans la méthode décrite par la suite par Rieke et al. en 1993[58], du 2,5-dibromo-3-alkylthiophène est traité par le « zinc de Rieke » hautement réactif[59] afin de former un mélange d'isomères organométalliques (figure 5, en bas). L'addition d'une quantité catalytique de Pd(PPh3)4 produit un polymère régioaléatoire, mais un traitement au Ni(dppe)Cl2 produit du PAT régiorégulier avec un rendement quantitatif[60].
Pour que les méthodes de McCullough et de Rieke produisent des PAT structurellement homogènes, elles nécessitent des températures basses, une déshydratation et une désoxygénation minutieuse, et des monomères bromés. De manière opposée, la polymérisation oxydative des thiophènes utilisant du chlorure ferrique décrite par Sugimoto en 1986 peut être réalisée à température ambiante sous des conditions moins exigeantes[61]. Cette méthode a prouvé son extrême « popularité ». Ainsi, le revêtement antistatique Baytron P de H.C. Stark est préparé à l'échelle industrielle en utilisant du chlorure ferrique (voir ci-dessous)[62].
De nombreuses études ont été menées pour tenter d'augmenter le rendement et la qualité du produit obtenu par technique de polymérisation oxydante. En plus du chlorure ferrique, d'autres agents oxydants, comme l'hydrate de chlorure ferrique, le perchlorate de cuivre et le perchlorate de fer ont été utilisés avec succès afin de polymériser le 2,2’-bithiophène[63]. Une addition lente de chlorure ferrique à la solution de monomère produit des poly(3-(4-octylphényl)thiophène)s avec un taux d'environ 94 % de H–T [32]. La précipitation du chlorure ferrique in situ (afin de maximiser la surface de réaction du catalyseur) donne des rendements et des conversions de monomères significativement supérieurs à l'addition directe du monomère au catalyseur cristallin[64],[65]. Des masses molaires plus importantes ont été obtenues quand de l'air sec a été « bullé » dans le mélange réactionnel lors de la polymérisation[66]. Une extraction de Soxhlet complète après une polymérisation avec des solvants polaires permet de fractionner de manière effective le polymère et d'enlever le catalyseur résiduel avant la spectroscopie RMN[29]. Utiliser un ratio catalyseur/monomère plus faible (2/1, au lieu de 4/1) peut permettre d'accroître la régiorégularité des poly(3-dodécylthiophène)s[67]. Andreani et al. ont indiqué de plus forts rendements pour les poly(dialkylterthiophène)s solubles dans le tétrachlorure de carbone plutôt que dans le chloroforme, qu'ils attribuent à la stabilité des espèces radicalaires dans le tétrachlorure de carbone[68]. Un catalyseur de qualité supérieure, additionné à taux plus faible et à température réduite, a été indiqué comme produisant des PAT de masse molaire plus importante sans résidu polymérique insoluble[69]. Laakso et al. ont utilisé un plan d'expérience pour déterminer que l'accroissement du taux catalyseur/monomère accroît le rendement de poly(3-octylthiophène), et annoncé qu'une durée de polymérisation plus importante augmentait aussi le rendement[70].
Le mécanisme de polymérisation oxydative utilisant le chlorure ferrique a été controversé. Sugimoto et al. ne spéculèrent aucun mécanisme dans leur publication de 1986[61]. En 1992, Niemi et al. ont proposé un mécanisme radicalaire, indiqué figure 6 (haut).
 Figure 6. Mécanismes proposés pour les polymérisations oxydatives par chlorure ferrique des thiophènes.
Figure 6. Mécanismes proposés pour les polymérisations oxydatives par chlorure ferrique des thiophènes.Ils ont basé leur mécanisme sur deux affirmations. Premièrement, parce qu'ils ont observé la polymérisation uniquement dans des solvants dans lesquels le catalyseur est soit partiellement soit complètement insoluble (chloroforme, toluène, tétrachlorure de carbone, pentane et hexane et pas dans le éther diéthylique, le xylène, l'acétone, ou l'acide formique), ils en ont conclu que les sites actifs de polymérisation devaient être localisés à la surface du chlorure ferrique solide. Donc, ils ont écarté les possibilités de réaction de deux cations radicalaires entre eux, ou de deux radicaux réagissant l'un avec l'autre, « parce que les ions chlorure à la surface du cristal empêcheraient des cations radicalaires ou des radicaux d'occuper des positions compatibles avec la dimérisation »[71]. Deuxièmement, l'utilisation du 3-methylthiophène comme monomère prototype, ils effectuèrent des calculs en mécanique quantique afin de déterminer les énergies et les charges atomiques totales sur les atomes de carbone sur les quatre espèces possibles de polymérisation (3-méthylthophène neutre, cation radicalaire, radical sur le carbone 2 et radical sur le carbone 5).
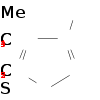 Sites réactionnels du 3-méthylthiophène
Sites réactionnels du 3-méthylthiophènePuisque le carbone le plus négatif sur le 3-méthylthiophene est le carbone 2, et que le carbone avec la plus forte population impaire d'électrons du cation radicalaire est aussi le carbone 2, ils conclurent que le mécanisme de cation radicalaire conduirait principalement à des liaisons 2–2, H–H. Ils calculèrent par la suite que les énergies totales des espèces avec des radicaux sur les carbones 2 et 5, et trouvèrent que les derniers étaient plus stables de 1,5 kJ/mol. Par conséquent, le radical le plus stable pourrait réagir avec les espèces neutres, formant des couplages tête-queue comme indiqué figure 6 (haut).
Andersson et al. ont proposé un mécanisme alternatif au cours de leurs études sur la polymérisation du 3-(4-octylphényl)thiophène avec du chlorure ferrique, dans laquelle ils obtinrent un haut degré de régiorégularité lorsque le catalyseur était ajouté lentement à la solution de monomères. Ils conclurent que, étant donné la sélectivité des couplages, et les conditions fortes d'oxydation, la réaction pourrait se faire via un mécanisme avec carbocation (voir figure 6, milieu)[32].
Le mécanisme radicalaire fut directement concurrencé dans une communication courte en 1995, lorsque Olinga et François notèrent que le thiophène pouvait être polymérisé par le chlorure ferrique dans l'acétonitrile, un solvant dans lequel le catalyseur est entièrement soluble. Leur analyse de la cinétique de la polymérisation du thiophène semblait aussi contredire les prédictions d'un mécanisme de polymérisation radicalaire[72]. Barbarella et al. étudièrent aussi l'oligomérisation des 3-(alkylsulfanyl)thiophènes, et conclurent d'après leurs calculs de chimie quantique, et à partir des considérations sur la stabilité induite du cation radicalaire lorsqu'il est délocalisé sur un oligomère plan conjugué, qu'un mécanisme par cation radicalaire analogue à celui généralement admis pour la polymérisation électrochimique était plus probable (figure 6, bas)[73]. Étant donnée la difficulté d'étudier un système avec un catalyseur hétérogène fortement oxydant qui rend difficile la caractérisation des polymères rigides, le mécanisme de polymérisation oxydante n'est pas encore déterminé. Cependant, le mécanisme par cation radicalaire indiqué dans la figure 6 est communément admis comme la voie de synthèse des PT la plus probable.
Applications
De nombreuses applications ont été proposées pour les PT conducteurs, y compris des transistors à effet de champ[74], des composés électroluminescents, des cellules photovoltaïques, des films photochimiques, matériaux d'optique non-linéaire[75], des piles électriques et diodes. En général, on peut considérer deux catégories d'applications pour les polymères conducteurs. Les applications statiques sont basées sur la conductivité électrique intrinsèque des matériaux, combinée avec la facilité de traitement et les propriétés communes aux polymères. Les applications dynamiques utilisent les modifications des propriétés optiques et de conduction dues soit à l'application de potentiels électriques, soit à des stimuli environnementaux.
 Figure 7. PEDOT-PSS (Baytron-P).
Figure 7. PEDOT-PSS (Baytron-P).On pourra citer comme exemple d'application statique le Baytron P à base de poly(3,4-éthylènedioxythiophène)-poly(styrène sulfonate) (PEDOT:PSS) (figure 7), utilisé de manière extensive comme revêtement antistatique (comme matériel d'emballage pour composants électronique, par exemple). AGFA recouvre ainsi 200 × 10 m de pellicule photographique avec du Baytron en raison de ses propriétés antistatiques. La fine couche de Baytron, a priori transparente et incolore, prévient les décharges électrostatiques durant le renroulage du film, et diminue le dépôt de poussière sur les négatifs après traitement.
Le PEDOT peut aussi être employé en application dynamique lorsqu'un potentiel est appliqué au film de polymère. Les propriétés électrochimiques du PEDOT sont utilisées pour produire des glaces et miroirs pouvant devenir opaques ou réflectifs lors de l'application d'un courant électrique[27]. Une application généralisée des fenêtres électrochromes pourrait faire économiser des milliards de dollars par an sur les coûts d'air conditionné[76]. Enfin, Phillips a commercialisé un téléphone portable avec un miroir PEDOT basculable électriquement (image).
Dans le domaine des cellules photovoltaïques en polymères, les structures à base de PT comme donneurs d'électrons (type p) et de fullerènes comme accepteurs d'électrons (type n) sont activement étudiées. Une jonction PEDOT:PSS / C60 peut faire l'affaire, mais les jonctions P3HT / PCBM sont de loin les plus prometteuses[77].
 Figure 8. PT ionosélectifs indiqués par Bäuerle (gauche) et Swager (droite).
Figure 8. PT ionosélectifs indiqués par Bäuerle (gauche) et Swager (droite).L'utilisation de PT comme capteurs répondant à une analyte a aussi été le sujet d'une recherche intense. En plus des applications de capteurs biologiques, les PT peuvent être aussi fonctionnalisés avec des récepteurs de synthèse pour détecter des ions métalliques ou aussi bien que des molécules chirales. Les PT avec chaînes latérales[78] ou principales[28] formant des éthers couronnes ont été signalés en 1993 par les équipes de recherche de Bäuerle et Swager, respectivement (figure 8). Des couches minces électrochimiquement polymérisées du PT avec éther couronne latéral de Bäuerle ont été exposés à des concentrations millimolaires de cations alcalins (Li, Na et K). Le courant passant au travers du film à potentiel fixe chute drastiquement dans les solutions d'ions lithium, beaucoup moins pour les solutions d'ions sodium et faiblement pour les solutions d'ions potassium. Les PT avec éthers couronnes en chaînes principales de Swager ont été préparés par couplage chimique et caractérisés par spectroscopie d'absorption. L'addition des mêmes cations alcalins conduit à des décalages d'absorption de 46 nm (Li), 91 nm (Na) et 22 nm (K). L'amplitude des déplacements correspond aux affinités de liaison ionique des éthers couronnes correspondants, résultant d'une torsion de la chaîne polymère conjuguée induite par la liaison ionique.
 Figure 9. PT chiral synthétisé par by Yashima et Goto.
Figure 9. PT chiral synthétisé par by Yashima et Goto.Au cours de leurs études des propriétés optiques des PT chiraux[79],[80],[81],[82], Yashima et Goto ont découvert qu'un PT avec une amine primaire chirale (figure 9) était sensible aux amino-alcools chiraux, produisant une division de l'image miroir dans les images réponses ICD dans la région de transition p[83]. Ce fut le premier exemple de reconnaissance chirale par les PT utilisant une méthode de détection chirale (spectroscopie DC). Ceci le distingue du travail précédemment effectué par Lemaire et al. qui utilisèrent une méthode achirale de détection (voltampérométrie cyclique) pour détecter l'incorporation de dopants anioniques chiraux dans un PT chiral polymérisé électrochimiquement[84].
Bibliographie
- (en) Handbook of Conducting Polymers (Eds: T. A. Skotheim, R. L. Elsenbaumer, J. R. Reynolds), Marcel Dekker, New York 1998. ISBN 0-8247-0050-3.
- (en) G. Schopf, G. Koßmehl, Polythiophenes: Electrically Conductive Polymers, Springer, Berlin 1997. ISBN 3-540-61483-4; ISBN 0-387-61483-4.
- (en) Synthetic Metals (journal). ISSN 0379-6779.
Sources
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Polythiophene » (voir la liste des auteurs)
Références
- [PDF]Street, G. B.; Clarke, T. C. IBM J. Res. Dev. 1981, 25, 51-57.
- Schopf, G.; Koßmehl, G. Adv. Polym. Sci. 1997, 129, 1–166.
- Roncali, J. Chem. Rev. 1992, 92, 711–738. DOI:10.1021/cr00012a009
- Roncali, J. Chem. Rev. 1997, 97, 173–205. DOI:10.1021/cr950257t
- McCullough, R. D. Adv. Mater. 1998, 10, 93-;116.
- Reddinger, J. L.; Reynolds, J. R. Adv. Polym. Sci. 1999, 145, 57–122.
- McQuade, D. T.; Pullen, A. E.; Swager, T. M. Chem. Rev. 2000, 100, 2537–2574. DOI:10.1021/cr9801014
- McCullough, R. D.; Tristramnagle, S.; Williams, S. P.; Lowe, R. D.; Jayaraman, M. J. Am. Chem. Soc. 1993, 115, 4910–4911. DOI:10.1021/ja00064a070
- Mastragostino, M.; Soddu, L. Electrochim. Acta 1990, 35, 463–466. DOI:10.1016/0013-4686(90)87029-2
- Loponen, M. T.; Taka, T.; Laakso, J.; Väkiparta, K.; Suuronen, K.; Valkeinen, P.; Österholm, J. E. Synth. Met. 1991, 41, 479–484. DOI:10.1016/0379-6779(91)91111-M
- Bartus, J. J. Macromol. Sci., Chem. 1991, A28, 917–924.
- Qiao, X. Y.; Wang, X. H.; Mo, Z. S. Synth. Met. 2001, 122, 449-454.
- McCarley, T. D.; Noble, C. O.; DuBois, C. J.; McCarley, R. L. Macromolecules 2001, 34, 7999–8004. DOI:10.1021/ma002140z
- Heffner, G. W.; Pearson, D. S. Synth. Met. 1991, 44, 341–347. DOI:10.1016/0379-6779(91)91821-Q
- Abdou, M. S. A.; Holdcroft, S. Synth. Met. 1993, 60, 93–96. DOI:10.1016/0379-6779(93)91226-R
- Rudge, A.; Raistrick, I.; Gottesfeld, S.; Ferraris, J. P. Electrochim. Acta 1994, 39, 273–287. DOI:10.1016/0013-4686(94)80063-4
- Bässler, H. Electronic Excitation. Dans Electronic Materials: The Oligomer Approach; Müllen, K.; Wegner, G., Eds.; Wiley-VCH: Weinheim, Germany 1998. ISBN 3-527-29438-4
- ten Hoeve, W.; Wynberg, H.; Havinga, E. E.; Meijer, E. W. J. Am. Chem. Soc. 1991, 113, 5887–5889. DOI:10.1021/ja00015a067
- Meier, H.; Stalmach, U.; Kolshorn, H. Acta Polym., 48, 379-384.
- Nakanishi, H.; Sumi, N.; Aso, Y.; Otsubo, T. J. Org. Chem. 1998, 63, 8632–8633. DOI:10.1021/jo981541y
- Izumi, T.; Kobashi, S.; Takimiya, K.; Aso, Y.; Otsubo, T. J. Am. Chem. Soc. 2003, 125, 5286–5287. DOI:10.1021/ja034333i
- de Souza, J. M.; Pereira, E. C. Synth. Met. 2001, 118, 167–170. DOI:10.1016/S0379-6779(00)00453-7
- Goto, H.; Yashima, E.; Okamoto, Y. Chirality 2000, 12, 396-399.
- Andersson, M.; Ekeblad, P. O.; Hjertberg, T.; Wennerström, O.; Inganäs, O. Polym. Commun. 1991, 32, 546–548.
- Nilsson, K. P. R.; Andersson, M. R.; Inganäs, O. J. Phys.: Condens. Matter 2002, 14, 10011-10020.
- Roux, C.; Leclerc, M. Macromolecules 1992, 25, 2141–2144. DOI:10.1021/ma00034a012
- Heuer, H. W.; Wehrmann, R.; Kirchmeyer, S. Adv. Funct. Mater. 2002, 12, 89-94.
- Marsella, M. J.; Swager, T. M. J. Am. Chem. Soc. 1993, 115, 12214–12215. DOI:10.1021/ja00078a090
- Barbarella, G.; Bongini, A.; Zambianchi, M. Macromolecules 1994, 27, 3039–3045. DOI:10.1021/ma00089a022
- Diaz-Quijada, G. A.; Pinto, B. M.; Holdcroft, S. Macromolecules 1996, 29, 5416–5421. DOI:10.1021/ma960126+
- Elsenbaumer, R. L.; Jen, K.-Y.; Miller, G. G.; Eckhardt, H.; Shacklette, L. W.; Jow, R. "Poly (alkyl thiophenes) and Poly (substituted heteroaromatic vinylenes): Versatile, Highly Conductive, Processible Polymers with Tunable Properties." In Electronic Properties of Conjugated Polymers (Eds: Kuzmany, H.; Mehring, M.; Roth, S.), Springer, Berlin, 1987. ISBN 0-387-18582-8
- Andersson, M. R.; Selse, D.; Berggren, M.; Järvinen, H.; Hjertberg, T.; Inganäs, O.; Wennerström, O.; Österholm, J. E. Macromolecules 1994, 27, 6503–6506. DOI:10.1021/ma00100a039
- Chen, T. A.; Wu, X.; Rieke, R. D. J. Am. Chem. Soc. 1995, 117, 233–244. DOI:10.1021/ja00106a027
- Xu, B.; Holdcroft, S. Macromolecules 1993, 26, 4457–4460. DOI:10.1021/ma00069a009
- Frommer, J. E. Acc. Chem. Res. 1986, 19, 2–9. DOI:10.1021/ar00121a001
- Elsenbaumer, R. L.; Jen, K. Y.; Oboodi, R. Synth. Met. 1986, 15, 169–174. DOI:10.1016/0379-6779(86)90020-2
- Hotta, S.; Rughooputh, S. D. D. V.; Heeger, A. J.; Wudl, F. Macromolecules 1987, 20, 212–215. DOI:10.1021/ma00167a038
- Hotta, S.; Soga, M.; Sonoda, N. Synth. Met. 1988, 26, 267–279. DOI:10.1016/0379-6779(88)90243-3
- Hotta, S. Synth. Met. 1987, 22, 103–113. DOI:10.1016/0379-6779(87)90528-5
- Hotta, S.; Rughooputh, S. D. D. V.; Heeger, A. J. Synth. Met. 1987, 22, 79–87. DOI:10.1016/0379-6779(87)90573-X
- Rughooputh, S. D. D. V.; Hotta, S.; Heeger, A. J.; Wudl, F. J. Polym. Sci., Polym. Phys. Ed. 1987, 25, 1071–1078.
- Patil, A. O.; Ikenoue, Y.; Wudl, F.; Heeger, A. J. J. Am. Chem. Soc. 1987, 109, 1858–1859. DOI:10.1021/ja00240a044
- Englebienne, P.; Weiland, M. Chem. Commun. 1996, 1651–1652. DOI:10.1039/cc9960001651
- Kim, B. S.; Chen, L.; Gong, J. P.; Osada, Y. Macromolecules 1999, 32, 3964–3969. DOI:10.1021/ma981848z
- Jung, S. D.; Hwang, D. H.; Zyung, T.; Kim, W. H.; Chittibabu, K. G.; Tripathy, S. K. Synth. Met. 1998, 98, 107-111.
- DeSimone, J. M.; Guan, Z.; Elsbernd, C. S. Science 1992, 257, 945–947.
- Li, L.; Counts, K. E.; Kurosawa, S.; Teja, A. S.; Collard, D. M. Adv. Mater. 2004, 16, 180-183.
- Murphy, A. R.; Fréchet, J. M. J.; Chang, P.; Lee, J.; Subramanian, V. J. Am. Chem. Soc. 2004, 126, 1596–1597. DOI:10.1021/ja0408193
- Lukkari, J.; Tuomala, R.; Ristimaki, S.; Kankare, J. Synth. Met. 1992, 47, 217-231.
- Lukkari, J.; Kankare, J.; Visy, C. Synth. Met. 1992, 48, 181-192.
- Lukkari, J.; Alanko, M.; Pitkänen, V.; Kleemola, K.; Kankare, J. J. Phys. Chem. 1994, 98, 8525-8535.
- Visy, C.; Lukkari, J.; Kankare, J. Synth. Met. 1997, 87, 81-87.
- Roncali, J.; Garreau, R.; Yassar, A.; Marque, P.; Garnier, F.; Lemaire, M. J. Phys. Chem. 1987, 91, 6706-6714.
- Meyer, V, . Ber. Deutsch. Chem. Ges. 1883, 16, 1465–1478.
- Yamamoto, T.; Sanechika, K.; Yamamoto, A. J. Polym. Sci., Polym. Lett. Ed. 1980, 18, 9–12.
- Lin, J. W. P.; Dudek, L. P. J. Polym. Sci., Polym. Chem. Ed. 1980, 18, 2869–2873.
- McCullough, R. D.; Lowe, R. D. J. Chem. Soc., Chem. Commun. 1992, 70–72.
- Chen, T. A.; O’Brien, R. A.; Rieke, R. D. Macromolecules 1993, 26, 3462–3463.DOI:10.1021/ma00065a036
- Zhu, L.; Wehmeyer, R. M.; Rieke, R. D. J. Org. Chem. 1991, 56, 1445–1453.
- Chen, T. A.; Rieke, R. D. J. Am. Chem. Soc. 1992, 114, 10087-10088.
- Sugimoto, R.; Taketa, S.; Gu, H. B.; Yoshino, K. Chem. Express 1986, 1, 635–638.
- Jonas, F.; Heywang, G.; Schmidtberg, W.; Heinze, J.; Dietrich, M. (en) Brevet U.S. 5,035,926, 1991.
- Ruckenstein, E.; Park, J. S. Synth. Met. 1991, 44, 293-306.
- Bizzarri, P. C.; Andreani, F.; Della Casa, C.; Lanzi, M.; Salatelli, E. Synth. Met. 1995, 75, 141-147.
- Fraleoni-Morgera, A.; Della Casa, C.; Lanzi, M.; Bizzarri, P. C. Macromolecules 2003, 36, 8617–8620. DOI:10.1021/ma0348730
- Pomerantz, M.; Tseng, J. J.; Zhu, H.; Sproull, S. J.; Reynolds, J. R.; Uitz, R.; Arnott, H. J.; Haider, M. I. Synth. Met. 1991, 41, 825-830.
- Qiao, X. Y.; Wang, X. H.; Zhao, X. J.; Liu, J.; Mo, Z. S. Synth. Met. 2000, 114, 261-265.
- Andreani, F.; Salatelli, E.; Lanzi, M. Polymer 1996, 37, 661-665.
- Gallazzi, M. C.; Bertarelli, C.; Montoneri, E. Synth. Met. 2002, 128, 91-95.
- Laakso, J.; Järvinen, H.; Skagerberg, B. Synth. Met. 1993, 55, 1204-1208.
- Niemi, V. M.; Knuuttila, P.; Österholm, J. E.; Korvola, J., Polymer 1992, 33, 1559-1562.
- Olinga, T.; François, B. Synth. Met. 1995, 69, 297-298.
- Barbarella, G.; Zambianchi, M.; DiToro, R.; Colonna, M.; Iarossi, D.; Goldoni, F.; Bongini, A. J. Org. Chem. 1996, 61, 8285-8292.
- Garnier, F. Field-Effect Transistors Based on Conjugated Materials. In Electronic Materials: The Oligomer Approach (Eds: Müllen, K.; Wegner, G.), Wiley-VCH, Weinheim, 1998. ISBN 3-527-29438-4
- Harrison, M. G.; Friend, R. H. Optical Applications. In Electronic Materials: The Oligomer Approach (Eds: Müllen, K.; Wegner, G.), Wiley-VCH, Weinheim, 1998. ISBN 3-527-29438-4
- Rosseinsky, D. R.; Mortimer, R. J. Adv. Mater. 2001, 13, 783-793.
- Mathieu Urien, Loïc Bailly, Laurence Vignau, Eric Cloutet, Anne De Cuendias, Guillaume Wantz, Henri Cramail, Lionel Hirsch, Jean-Paul Parneix, « Effect of the regioregularity of poly(3-hexylthiophene) on the performance of organic photovoltic devices », dans Polymer International, vol. 57, no 5, 2008, p. 764-769 [texte intégral, lien DOI (pages consultées le 12/06/2009)]
- Bäuerle, P.; Scheib, S. Adv. Mater. 1993, 5, 848-853.
- Goto, H.; Okamoto, Y.; Yashima, E. Chem. Eur. J. 2002, 8, 4027-4036.
- Goto, H.; Okamoto, Y.; Yashima, E. Macromolecules 2002, 35, 4590–4601.
- Goto, H.; Yashima, E. J. Am. Chem. Soc. 2002, 124, 7943-7949.
- Sakurai, S.; Goto, H.; Yashima, E. Org. Lett. 2001, 3, 2379-2382.
- Yashima, E.; Goto, H.; Okamoto, Y. Macromolecules 1999, 32, 7942–7945.
- Lemaire, M.; Delabouglise, D.; Garreau, R.; Guy, A.; Roncali, J., J. Chem. Soc., Chem. Commun. 1988, 658–661.

La version du 28 avril 2007 de cet article a été reconnue comme « bon article », c'est-à-dire qu'elle répond à des critères de qualité concernant le style, la clarté, la pertinence, la citation des sources et l'illustration.

{kind=link}
Wikimedia Foundation. 2010.