- Polymère en solution
-
Comprendre le comportement des polymères en solution est important pour certaines applications comme les gels et crèmes cosmétiques. Il y a possibilité de contrôler la viscosité du produit obtenu et d'éviter une éventuelle démixtion (séparation de phase). On constate par exemple que le styrène est miscible en toute proportion dans le cyclohexane à 20 °C, mais que lorsqu'on polymérise le styrène, il n'y a plus miscibilité. Différentes théories peuvent expliquer ces phénomènes.
Sommaire
Théorie de Flory-Huggins
On modélise une chaîne de polymère par un ensemble de N maillons (unités monomères) de volume a. Le nombre moyen de chaînons est lié à au degré de polymérisation et à la masse molaire du polymère. Il est en solution dans un solvant avec une fraction volumique ou une concentration connue.
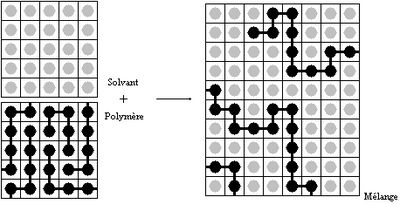
Mélange polymère solvant
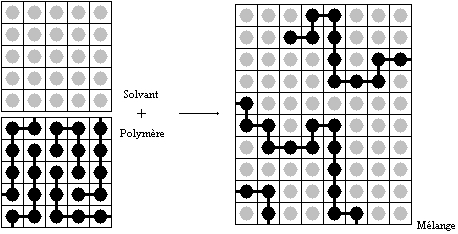
On fait l'hypothèse que le volume molaire des unités monomères est égal à celui des molécules de solvant. Le mélange peut être représenté comme ceci:
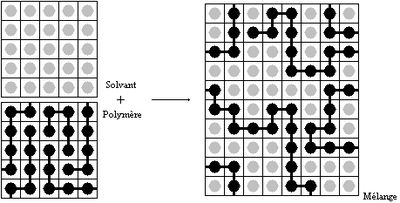 Réseau de Flory-Huggins représentant le polymère en solution
Réseau de Flory-Huggins représentant le polymère en solution
À température et pression fixées, l'enthalpie libre de mélange doit être négative pour que le mélange ait lieu. Dans le cas d'une solution régulière, des calculs de thermodynamique statistique permettent d'estimer cette enthalpie libre:
 avec kB la constante de Boltzmann, T la température en kelvins, ϕ la fraction volumique et χ un paramètre sans dimension
avec kB la constante de Boltzmann, T la température en kelvins, ϕ la fraction volumique et χ un paramètre sans dimension
Pour un polymère dans un solvant, Flory a posé[1]: avec N le nombre moyen d'unités monomère par chaîne.
avec N le nombre moyen d'unités monomère par chaîne.Cette enthalpie libre est en fait la somme de deux termes[2]
 :
:  est l'enthalpie de mélange et
est l'enthalpie de mélange et  l'entropie de mélange, c'est cette dernière qui est diminuée par le facteur N. Quand N ou la masse molaire (c'est équivalent) augmente, l'entropie diminue, donc le désordre également. Il y a en effet moins de possibilités de répartir les unités monomères et les molécules de solvant sur la grille ci dessus quand les chaînes sont plus longues. Il en résulte une enthalpie libre de mélange plus difficilement négative quand le polymère a une plus forte masse molaire. Ceci est en accord avec la précipitation qu'on peut observer quand on synthétise un polymère en solution, donc dans un solvant.
l'entropie de mélange, c'est cette dernière qui est diminuée par le facteur N. Quand N ou la masse molaire (c'est équivalent) augmente, l'entropie diminue, donc le désordre également. Il y a en effet moins de possibilités de répartir les unités monomères et les molécules de solvant sur la grille ci dessus quand les chaînes sont plus longues. Il en résulte une enthalpie libre de mélange plus difficilement négative quand le polymère a une plus forte masse molaire. Ceci est en accord avec la précipitation qu'on peut observer quand on synthétise un polymère en solution, donc dans un solvant.Paramètre de Flory
Le paramètre χ qui intervient dans l'expression de l'enthalpie et donc dans celle de l'enthalpie libre, permet de tenir compte des interactions entre le polymère et le solvant. Dans ce modèle[3],[4],
 où Z est le nombre de premiers voisins des unités monomères ou des molécules de solvant,
où Z est le nombre de premiers voisins des unités monomères ou des molécules de solvant,  l'énergie d'interaction entre unité monomère et solvant,
l'énergie d'interaction entre unité monomère et solvant,  l'énergie d'interaction entre unités monomères, et
l'énergie d'interaction entre unités monomères, et  l'énergie d'interaction entre molécules de solvant.
l'énergie d'interaction entre molécules de solvant.
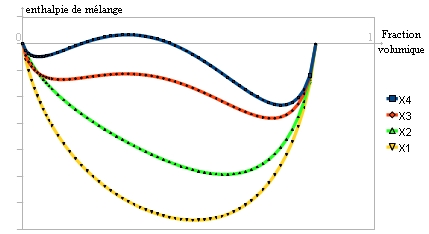
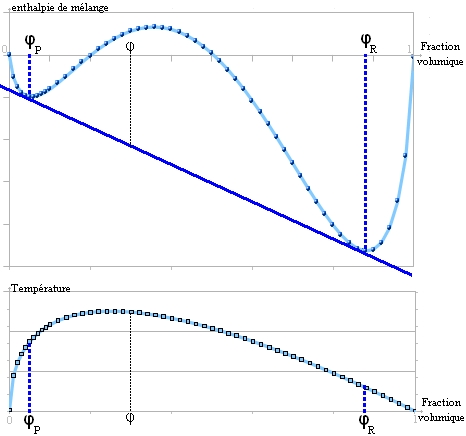
Il s'agit du paramètre de Flory, qui joue un rôle important dans l'évolution de l'enthalpie libre: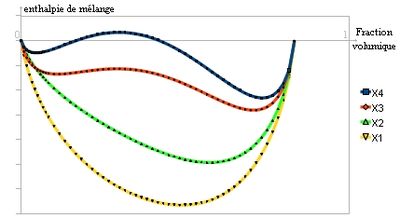 Lorsque le paramètre de Flory augmente: χ1 < χc < χ2 < χ3 < χ4, une zone de démixtion apparaît et couvre une plage de fractions volumiques de plus en plus grande.
Lorsque le paramètre de Flory augmente: χ1 < χc < χ2 < χ3 < χ4, une zone de démixtion apparaît et couvre une plage de fractions volumiques de plus en plus grande.À partir d'une certaine valeur de χ, la courbe présente deux points d'inflexion et il est possible de tracer une double tangente[5]. C'est le signe d'une séparation en deux phases comme ceci[1]:
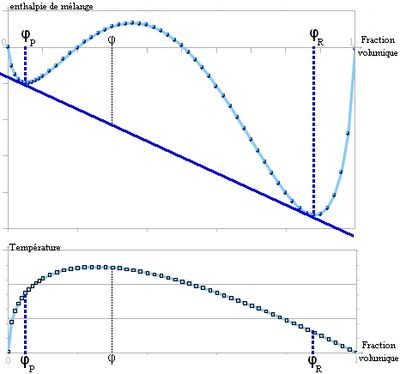 Il y a démixtion en 2 phases : l'une riche en polymère et l'autre pauvre, les fractions volumiques en polymère des deux phases sont ϕR et ϕP respectivement.
Il y a démixtion en 2 phases : l'une riche en polymère et l'autre pauvre, les fractions volumiques en polymère des deux phases sont ϕR et ϕP respectivement.On peut calculer cette valeur critique de paramètre de Flory[1], notée
 et obtenue pour
et obtenue pour  .
.Pour résumer, la démixtion apparait quand le paramètre de Flory dépasse une valeur critique qui dépend de la masse molaire. Ceci peut se comprendre également en comparant les interactions qui apparaissent dans l'expression de ce paramètre. Lorsque χ est grand,
domine. Cette énergie attractive est bien sur négative et lorsqu'elle est grande, elle diminue en valeur absolue : l'attraction entre polymère et solvant faiblit. Les maillons des chaînes de polymère sont attirés entre eux et le polymère sera de moins en moins solubilisé.On peut accéder à ce paramètre de Flory expérimentalement par des mesures de pression osmotique par exemple.
Diagramme d'état φ-T
Pour un système polymère/solvant, si les interactions de VdW de type London dominent (typiquement en milieu apolaire), on peut montrer que χ varie comme l'inverse de la température. À basse température le paramètre de Flory sera grand et on aura une démixtion, et inversement.
Par contre lorsque les liaisons H dominent (milieu polaire), χ augmente avec la température et on a donc la situation inverse. À haute température l'agitation thermique rompt les liaisons H qui liaient polymère au solvant et la séparation de phase a lieu.
Pour obtenir ces diagrammes de phase, on peut partir de tubes contenant différentes fractions molaires en polymère et observer à quel moment chaque tube devient trouble lorsqu'on les refroidit à partir d'une température pour laquelle on a mélange[6]. Le trouble est dû à la diffusion de la lumière dans toutes les directions par les particules qui apparaissent quand la séparation de phase se produit : Apparition du trouble dû aux gouttelettes qui diffusent la lumière
Apparition du trouble dû aux gouttelettes qui diffusent la lumièreApproche de Hildebrand, paramètre de solubilité
On définit l'énergie de cohésion du système et la densité d'énergie cohésive qui s'obtient en divisant la première par le volume molaire[6]:
 . Le paramètre de solubilité est alors égal à la racine carrée de cette densité c[6]:
. Le paramètre de solubilité est alors égal à la racine carrée de cette densité c[6]:  . Les valeurs de paramètre de solubilité sont tabulées pour de nombreux solvants et de nombreux polymères[7]. on peut montrer que le paramètre de Flory vu précédemment peut s'écrire[8]:
. Les valeurs de paramètre de solubilité sont tabulées pour de nombreux solvants et de nombreux polymères[7]. on peut montrer que le paramètre de Flory vu précédemment peut s'écrire[8]:
Avec R la constante des gaz parfaits. Si on cherche un bon solvant pour un polymère donné il faut donc chercher un paramètre de solubilité qui soit proche de celui du polymère, pour minimiser le paramètre de Flory. On peut ainsi éliminer bon nombre de solvants pour un polymère donné, mais même si les paramètres de Hildebrand sont proches la solubilité n'est pas assurée[9].
On peut aussi calculer le paramètre de solubilité comme ceci[5]:
 où G(i) est la constante d'attraction molaire du groupe i[7].
où G(i) est la constante d'attraction molaire du groupe i[7].Pression osmotique et qualité de solvant
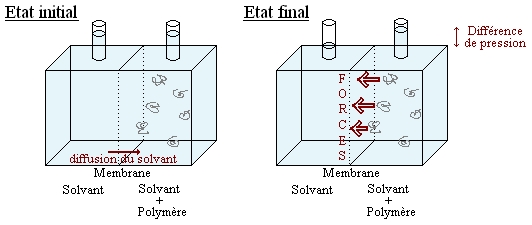
Soient deux compartiments séparés par une membrane semi perméable:
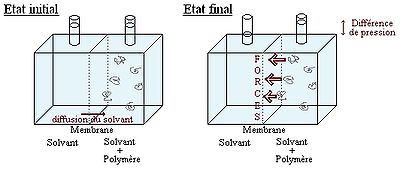 Illustration de la pression osmotique
Illustration de la pression osmotiqueLe compartiment de droite contient le polymère en solution dans un solvant, tandis que le compartiment de gauche ne contient que le solvant. On observe une variation de la pression entre les 2 compartiments. Plus précisément les chaînes de polymère exercent une force sur la paroi. Cette force par unité de surface est appelée pression osmotique. On peut la calculer moyennant quelques hypothèses à partir de l'enthalpie de mélange vu précédemment.
Si la fraction volumique en polymère n'est pas trop grande, on obtient:
On se rend compte que la valeur du paramètre de Flory joue ici encore un rôle:
- s'il est supérieur à 1/2, la pression osmotique diminue, cela revient à dire qu'il y a moins de chocs de particules sur la paroi et qu'elles ont plutôt tendance à rester entre elles. le polymère est plus difficile à dissoudre, on est en régime mauvais solvant. Le polymère peut cependant être dissous ou avoir précipité : χC > 1 / 2.
- s'il est inférieur à 1/2, la pression osmotique augmente et c'est l'inverse : le solvant dissout bien le polymère et on dit qu'on est en régime bon solvant. Si le polymère est sous forme de pelotes, cela se traduira par un gonflement de ces dernières[6]. Le monomère est souvent un bon solvant pour son polymère, comme par exemple le styrène pour le polystyrène.
- la valeur limite χ = 1 / 2 correspond à ce qu'on appelle solvant θ (théta), auquel correspond une température θ[6]. La température critique Tc vue plus haut tend vers cette température θ lorsque la masse molaire tend vers l'infini[10], ce qui permet d'accéder à θ par extrapolation.
Le paramètre de Flory χ varie avec la température. Lorsqu'il prend la valeur limite 1/2, la température associée porte le nom de température théta. Cette température particulière peut souvent se trouver dans des tables pour un couple polymère/solvant. Par exemple le cyclohexane est un solvant théta du polystyrène[5] à 307,2 K.
Théorie de Flory-Krigbaum
Cette théorie introduit la notion de volume exclu : il y a une répulsion entre chaînes, de nature géométrique, qui interdit la présence du centre de masse d'une autre chaîne de polymère autour d'une chaîne donnée.
 Modélisation de la répartition des chaînes en tenant compte du volume exclu
Modélisation de la répartition des chaînes en tenant compte du volume excluLe paramètre de Flory calculé précédemment est modifié[11]:
 . À un couple polymère - solvant on peut associer une température θ et un paramètre ψ dit paramètre entropique, tous deux indépendants de la température[11]:
. À un couple polymère - solvant on peut associer une température θ et un paramètre ψ dit paramètre entropique, tous deux indépendants de la température[11]:  . Connaissant la température critique Tc (d'apparition du trouble) pour des échantillons d'un polymère de différentes masses molaires, on peut déterminer graphiquement la température θ et le paramètre entropique.
. Connaissant la température critique Tc (d'apparition du trouble) pour des échantillons d'un polymère de différentes masses molaires, on peut déterminer graphiquement la température θ et le paramètre entropique.Le volume exclu (u), le paramètre de Flory et le second coefficient de Viriel du couple polymère/solvant A2 sont liés par les relations suivantes:
 . Na est le nombre d'avogadro, N le nombre de segments, M la masse molaire, a le volume d'une unité monomère. Lorsque le paramètre de Flory est inférieur à 0.5, le volume exclu augmente : il y a extension des chaînes et on se situe en régime bon solvant. En mauvais solvant c'est l'inverse, il y a contraction des chaînes. On peut obtenir expérimentalement le volume exclu par osmométrie[12].
. Na est le nombre d'avogadro, N le nombre de segments, M la masse molaire, a le volume d'une unité monomère. Lorsque le paramètre de Flory est inférieur à 0.5, le volume exclu augmente : il y a extension des chaînes et on se situe en régime bon solvant. En mauvais solvant c'est l'inverse, il y a contraction des chaînes. On peut obtenir expérimentalement le volume exclu par osmométrie[12].Régimes de concentration et loi d'échelle
Il existe différents régimes, selon la concentration et la masse du polymère en solution. Pour des raisons entropiques, les chaînes de polymère ne sont pas dépliées mais elles se contractent sur elles-mêmes pour former des pelotes.
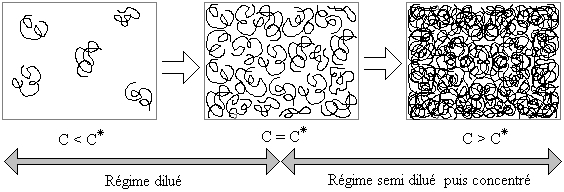
Du régime dilué au régime semi dilué
Soient des pelotes de polymère en solution. Lorsqu'on augmente la concentration (ou la fraction volumique ce qui revient au même), la distance entre pelotes diminue. À partir d'un moment, il y a contact entre ces dernières et on appelle concentration critique de recouvrement la concentration associée. Elle marque la transition entre le régime dilué et le régime semi dilué. Il est encore possible d'augmenter la concentration dans le régime semi dilué, car la fraction de polymère contenue dans une pelote est vraiment faible.
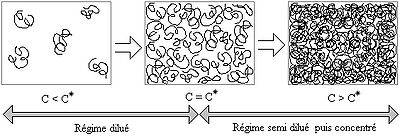 Effet d'une augmentation de la concentration sur les pelotes de polymère en solution
Effet d'une augmentation de la concentration sur les pelotes de polymère en solutionOn peut calculer la concentration critique de recouvrement à l'aide de la relation suivante[3],[13]:
 .
.
La fraction volumique dépend du nombre de segments des chaînes à la puissance -4/5.Rayon de giration en régime dilué
Cette distance a une influence sur les propriétés physicochimiques du système. En bon solvant[6],
 , avec Rg le rayon de giration des pelotes, N le nombre de segments de la chaîne qui constitue la pelote et a la taille de ces segments. En solvant θ,
, avec Rg le rayon de giration des pelotes, N le nombre de segments de la chaîne qui constitue la pelote et a la taille de ces segments. En solvant θ,  . On peut comprendre cette différence d'exposant en prenant en compte le fait qu'en bon solvant χ < 1 / 2, les interactions répulsives entre segments de pelotes dominent. Les pelotes gonflent et leur rayon de giration augmente.
. On peut comprendre cette différence d'exposant en prenant en compte le fait qu'en bon solvant χ < 1 / 2, les interactions répulsives entre segments de pelotes dominent. Les pelotes gonflent et leur rayon de giration augmente.
En solvant θ, il existe différents modèles pour calculer le rayon de giration:- modèle de la marche au hasard[2]: < Rg > 2 = N.a2
- rotation libre[6],[14]:
 où θ est l'angle de valence, pris selon cette convention:
où θ est l'angle de valence, pris selon cette convention:

- rotation gênée: on tient compte des interactions entre groupements moléculaires.
 . ϕ est l'angle de rotation interne ou angle dièdre. On peut voir cos(ϕ) comme un coefficient de rigidité.
. ϕ est l'angle de rotation interne ou angle dièdre. On peut voir cos(ϕ) comme un coefficient de rigidité.
Si on n'est plus en conditions θ, on peut introduire un coefficient d'expansion volumique comme ceci[15]:
 .
.Grandeur caractéristique en régime semi dilué
Lorsque les pelotes s'interpénètrent, on ne peut plus vraiment parler de rayon de giration, vu qu'on ne peut pas distinguer les pelotes. On définit la distance moyenne entre points d'enchevêtrement, notée ξ. Cette grandeur est égale à Rg lorsqu'on est à la concentration critique de recouvrement. Elle diminue ensuite lorsque la concentration augmente. Les propriétés du système dépendent de cette grandeur dans le régime semi dilué. On peut montrer que[2]:
 en bon solvant.
en bon solvant. ξ est une longueur de corrélation, ou maille de réseau.
ξ est une longueur de corrélation, ou maille de réseau.Le régime enchevêtré
Il est possible quand la masse dépasse une masse critique d'enchevêtrement Mc. Il peut apparaître dans le régime semi dilué, voire dans le régime concentré (quand on augmente encore plus la concentration). Dans le régime enchevêtré, le mouvement des chaînes est très gêné par celui des autres chaînes. Son mouvement est décrit par le modèle de reptation. La viscosité dépend alors de la concentration à la puissance 3.75 et de la masse au cube. En régime non enchevêtré, la viscosité dépend de la concentration à la puissance 2.5 seulement et les chaînes ont une dynamique dite de Rouse. Cette effet plus ou moins grand de la concentration permet de jouer sur la viscosité du système de manière importante et peut être utilisé lors de la confection de gels de polymère.
Analyse des polymères en solution
Il existe différentes techniques pour étudier les polymères en solution.
- méthodes thermodynamiques colligatives :
- osmométrie: accès à Mn[5], au deuxième coefficient de viriel A2 ;
- tonométrie ;
- cryoscopie et ébulliométrie.
- méthodes viscosimétriques :
- accès à la viscosité, à la masse molaire moyenne viscosimétrique[6] (Mn<Mv<Mw), au rayon de giration des pelotes.
- techniques hydrodynamiques:
- CES, accès aux masses molaires moyennes en masse et en moles.
- techniques de diffusion de lumière :
- diffusion statique de la lumière: accès à Rg, Mw[6], A2 ;
- diffusion dynamique de la lumière: accès au rayon hydrodynamique et au coefficient de diffusion.
Voir aussi
Références
- Menno A. van Dijk et André Wakker, Concepts of polymer thermodynamics, CRC Press, 1997, 209 p. (ISBN 1566766230, 9781566766234) [lire en ligne]
- D Espinat, Application des techniques de diffusion de la lumière, des rayons X et des neutrons à l'étude des systèmes colloïdaux, Éditions TECHNIP, 1992, 131 p. (ISBN 2710806177, 9782710806172) [lire en ligne]
- Hans-Georg Elias, Macromolecules: Volume 3: Physical Structures and Properties, Wiley-VCH, 2008, 699 p. (ISBN 3527311742, 9783527311743) [lire en ligne]
- Certains auteurs le définissent avec Z-2 plutôt que Z
- J. M. G. Cowie, Polymers: chemistry and physics of modern materials, CRC Press, 1991, 436 p. (ISBN 0748740732, 9780748740734) [lire en ligne]
- Hans-Henning Kausch, Nicole Heymans, Christopher-John Plummer et Pierre Decroly, Matériaux polymères : propriétés mécaniques et physiques, [principes de mise en oeuvre], PPUR presses polytechniques, 2001, 657 p. (ISBN 2880744156, 9782880744151) [lire en ligne]
- Valeurs de paramètre de hildebrand et de constantes d'attractions molaires
- Jean Vidal, Thermodynamique: application au génie chimique et à l'industrie pétrolière, Éditions TECHNIP, 1997, 500 p. (ISBN 2710807157, 9782710807155) [lire en ligne]
- Andreĭ Aleksandrovich Askadskiĭ, Physical properties of polymers: prediction and control, CRC Press, 1996, 336 p. (ISBN 2884492208, 9782884492201) [lire en ligne]
- Michio Kurata, Thermodynamics of polymer solutions, Taylor & Francis, 1982, 294 p. (ISBN 3718600234, 9783718600236) [lire en ligne]
- Yuri S. Lipatov et Anatoly E. Nesterov, Thermodynamics of polymer blends, CRC Press, 1997, 450 p. (ISBN 1566766249, 9781566766241) [lire en ligne]
- Paul C. Hiemenz, Polymer chemistry: the basic concepts, CRC Press, 1984, 738 p. (ISBN 082477082X, 9780824770822) [lire en ligne]
- Certains auteurs la définissent avec un coefficient multiplicatif 0.74 au numérateur
- Si on prend une autre convention pour définir l'angle, la formule est inversée
- cours en anglais
Wikimedia Foundation. 2010.