- Thermodynamique statistique
-
Physique statistique
La physique statistique a pour but d'expliquer le comportement et l'évolution de systèmes physiques comportant un grand nombre de particules (on parle de systèmes macroscopiques), à partir des caractéristiques de leurs constituants microscopiques (les particules). Ces constituants peuvent être des atomes, des molécules, des ions, des électrons, des photons, des neutrinos, ou des particules élémentaires. Ces constituants, et les interactions qu'ils peuvent avoir entre eux, sont en général décrits par la mécanique quantique, mais la description macroscopique d'un ensemble de tels constituants ne fait, elle, pas directement appel (ou en tout cas pas toujours) à la mécanique quantique. De fait, cette description macroscopique, en particulier la thermodynamique, a été obtenue pour partie avant le développement de la mécanique quantique en tant que théorie physique, essentiellement dans la seconde moitié du XIXe siècle.
On distingue la physique statistique d'équilibre (au sens d'équilibre thermodynamique), auquel cet article est consacré, de la physique statistique hors d'équilibre.
Sommaire
Historique
La physique statistique (appelé aussi « thermodynamique statistique ») fut introduite initialement sous la forme de la théorie cinétique des gaz à partir du milieu du XIXe siècle, principalement par Kelvin, Maxwell et Boltzmann. Cette première approche visait à proposer un modèle simple de la matière à l'échelle atomique, et en particulier des collisions entre atomes ou molécules, pour reproduire le comportement de certaines quantités macroscopiques. C'est à cette époque que l'interprétation de la pression comme mesure de la quantité de mouvement des constituants d'un gaz a été formalisée.
La mécanique statistique fut formalisée en 1902 par Gibbs[1], son formalisme permettant de généraliser et de justifier a posteriori les principes de la thermodynamique d'équilibre.
Les premières extensions de la physique statistique, par rapport à la mécanique statistique, ont été l'introduction des propriétés électriques et magnétiques de la matière au sein des modèles, permettant la description des transitions de phase dans les matériaux magnétiques ou diélectriques, comme la transition ferromagnétique.
Une autre étape importante fut la modification des formules statistiques, entre les années 1920 et 1930, pour tenir compte des effets de l'indiscernabilité au niveau quantique des particules (principe d'exclusion de Pauli). Cette modification fut effectuée par Bose et Einstein pour les systèmes de particules de spin entier (bosons) et par Fermi et Dirac pour les systèmes de particules de spin demi-entier (fermions).
Introduction et généralités
Typiquement, à notre échelle, un système matériel à l'équilibre thermodynamique est décrit à l'aide d'un nombre restreint de paramètres dits macroscopiques caractérisant les valeurs de certaines grandeurs physiques macroscopiques ; par exemple, un volume de gaz à l'équilibre est caractérisé par sa densité μ, sa pression P, sa température T, et son volume V. La physique statistique établit des liens statistiques entre ces grandeurs physiques macroscopiques et d'autres grandeurs microscopiques caractérisant les constituants élémentaires (atomes ou molécules) du système matériel.
Cette procédure est utile, car il est en pratique impossible de suivre l'évolution des constituants individuels du gaz. Par exemple, un litre d'air contient environ 3×1022 molécules (un nombre de l'ordre du nombre d'Avogadro). Même s'il était possible de suivre l'évolution individuelles de chacune d'elles, cela ne donnerait pas d'information pertinente sur le système. Le but de la physique statistique est de définir les quantités macroscopiques pertinentes qui permettent de décrire un tel système composé d'un très grand nombre de particules, et les relations entre ces différentes quantités. Elle doit aussi relier ces quantités macroscopiques à des quantités d'origine microscopiques qui décrivent les constituants du système. Par exemple, la température d'un gaz parfait est une mesure de l'énergie cinétique totale des particules qui le composent.
Une des raisons qui permet un passage du microscopique au macroscopique est en fait le nombre gigantesque des constituants du système. Pour cette raison, on parle de l'énergie cinétique E totale des molécules d'un gaz, car les fluctuations statistiques ΔE d'une telle quantité (il est possible que l'énergie cinétique totale des molécules du gaz soit, dans une petite région, légèrement différente de la valeur moyenne attendue) sont limitées par la relation (issue de la loi de Poisson)
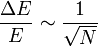 ,
,
où N est le nombre de molécules du volume de gaz considéré. Même pour des petits volumes, par exemple de l'ordre de 1 mm3 pour un gaz à TPN, soit environ 3×1016 particules, les fluctuations de l'énergie cinétique totale sont inférieures à 10-8 et en pratique presque toujours négligeables.
Postulat fondamental
Énoncé
Le postulat fondamental de la physique statistique d'équilibre (aussi connu comme le postulat des probabilités a priori égales) est:
Étant donné un système isolé en équilibre, il se trouve avec probabilités égales dans chacun de ses micro-états accessibles. Ce postulat est une hypothèse fondamentale en physique statistique : il signifie qu'un système n'a pas de préférence pour n'importe quel de ses microétats accessibles. Étant donnés Ω microétats à énergie donnée, la probabilité que le système se trouve à un microétat particulier est p = 1/Ω. Ce postulat, nécessaire, permet de conclure que pour un système à l'équilibre, l'état thermodynamique (le macroétat) qui peut résulter du plus grand nombre de microétats est aussi le macroétat le plus probable du système.
Par rapport aux postulats sous-tendant la théorie cinétique des gaz, il s'agit d'un saut dans l'abstraction qui permet toutes les généralisations. Cet énoncé remplace avantageusement les modèles microscopiques à la fois simplistes et pourtant lourdement calculatoires par des incertitudes statistiques. Le modèle s'applique a priori à tout système possédant un mécanisme de redistribution statistique de l'énergie au niveau microscopique.
Origine : l'hypothèse ergodique
Dans le cadre naissant de la théorie cinétique des gaz, Boltzmann a formulé en 1871 une hypothèse, connue aujourd'hui sous le nom d'hypothèse ergodique : « Le point représentatif d'un système hamiltonien invariant par translation dans le temps passe au cours du temps par chaque point de l'hypersurface d'énergie constante. »
Il a été prouvé en 1910 que cette hypothèse était fausse, mais on a depuis démontré que certains systèmes physiques vérifient l'hypothèse quasi-ergodique :
Le point représentatif d'un système hamiltonien invariant par translation dans le temps passe au cours du temps aussi près que l'on veut de chaque point de l'hypersurface d'énergie constante. Cependant, en dépit de progrès très important réalisés en théorie ergodique et en théorie du chaos, l'utilisation de l'hypothèse quasi-ergodique pour justifier le postulat fondamental de la physique statistique reste à ce jour controversée [2].
Équiprobabilité et information
En théorie de l'information, l'information est l'opposée du logarithme de la probabilité :
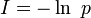 .
.Dans ce contexte, l'entropie est définie comme la moyenne de l'information contenue dans le système :
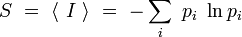
C'est une fonction continue de plusieurs variables, dont on peut montrer qu'elle admet un maximum global lorsque tous les pi sont égaux. Ceci signifie qu'avec l'hypothèse d'équiprobabilité, S est maximale. On interprète cela en disant qu'on a alors un minimum d'information, ou encore un maximum d'incertitude, sur le système.
Par contre, quand l'un des évènements est certain, sa probabilité vaut 1 tandis que tous les autres pi sont nuls. On connait alors exactement la configuration du système et l'incertitude est nulle. Dans ce cas l'entropie admet sa valeur minimale, en l'occurence 0.
Généralisations aux problèmes analogues
Les méthodes de description mathématique développées dans le cadre de la physique statistique ont trouvé des applications dans pratiquement tous les domaines de la physique moderne ou de la chimie, mais aussi en économie, dans les sciences humaines, etc.
Outils et procédés
La formulation moderne de cette théorie se base sur la description des systèmes physiques étudiés par le biais d'ensembles statistiques. De tels ensembles représentent la totalité des configurations possibles du système associées à leur probabilités de réalisation. À chaque ensemble est associée une fonction de partition qui, par manipulations mathématiques, permet d'extraire les grandeurs thermodynamiques du système. Selon les relations du système avec le reste de l'univers, on distingue généralement trois types d'ensemble, du plus simple au plus complexe :
- l'ensemble microcanonique
- l'ensemble canonique
- l'ensemble grand canonique
Tableau résumant
les ensembles
en physique statistiqueEnsembles Microcanonique Canonique Grand-canonique Variables indépendantes E, N, V ou B T, N, V ou B T, μ, V ou B Fonction microscopique nombre des micro-états
ΩFonction de partition canonique
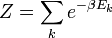
Fonction de partition grand-canonique
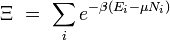
Potentiel thermodynamique Entropie
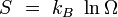
Energie libre
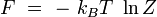
Grand potentiel
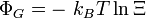
Ensemble microcanonique
Cet ensemble décrit le cas idéal d'un système complètement isolé d'énergie E constante, et n'échangeant donc ni particule, ni énergie, ni volume avec le reste de l'univers. L'intérêt de ce modèle est qu'il permet de définir l'entropie sous sa forme la plus simple.
Entropie microcanonique
Le système étant à l'équilibre macroscopique, mais libre d'évoluer à l'échelle microscopique entre Ω micro-états différents, son entropie est donnée par la formule de Boltzmann (1877) :
où kB est la constante de Boltzmann. Cette définition correspond à l'entropie de Shannon :
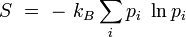
d'une configuration de Ω micro-états équiprobables :
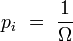
Extensivité de l'entropie
L'énergie macroscopique totale E du système isolé étudié est toujours supérieure ou égale à une certaine valeur E0 minimale, appelée énergie de l'état fondamental du système. De plus, le comptage du nombre Ω de micro-états du système fermé d'énergie totale E nécessite[3] en général l'introduction d'une certaine incertitude ΔE d'ordre mésoscopique.
On peut montrer que, pour un système « ordinaire »[4], le nombre Ω de micro-états est une fonction rapidement croissante de l'énergie de la forme :
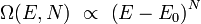
où N le nombre de degrés de liberté total du système, supposé très grand. Il est alors possible de montrer[5] que lorsque l'énergie totale E n'est pas trop proche de sa valeur minimale, l'entropie S(E,N) calculée par la formule de Boltzmann est :
- de l'ordre de N, donc l'entropie microcanonique est bien extensive à la limite thermodynamique.
- indépendante de la valeur exacte de l'incertitude ΔE.
Ensemble canonique
L'ensemble canonique décrit un système fermé en équilibre thermique avec un thermostat extérieur. Ce système fermé peut donc échanger de l'énergie sous forme de transfert thermique avec l'extérieur, à l'exclusion de toute autre quantité.
Fonction de partition canonique
Dans les conditions citées ci-dessus, on démontre que la probabilité pi pour que le système fermé réalise un état i d'énergie Ei est donnée par la formule de Boltzmann :
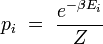
où le facteur
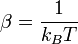 , parfois appelé « température inverse », traduit la thermalisation du système avec le thermostat extérieur à la température T. Le facteur de normalisation Z se calcule en écrivant la condition des probabilités totales :
, parfois appelé « température inverse », traduit la thermalisation du système avec le thermostat extérieur à la température T. Le facteur de normalisation Z se calcule en écrivant la condition des probabilités totales :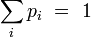
Z est appelé la fonction de partition canonique du système fermé, et s'écrit explicitement :

Cette fonction de partition permet de déduire toutes les grandeurs macroscopiques du système fermé comme moyennes des grandeurs microscopiques associées.
Observables macroscopiques
L'énergie interne U est la moyenne macroscopique de l'ensemble des énergies microscopiques Ei :
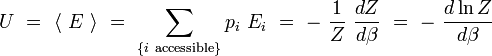
De même, pour toute grandeur A prenant des valeurs Ai definies sur les micros états i associés aux énergies Ei, on peut définir la valeur moyenne :
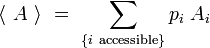
Appliquons en particulier cette formule à l'entropie, en posant que les micros-états Ei définissent des systèmes représentables comme des ensembles microcanoniques, nous avons défini pour chaque micro-état i une entropie microcanonique :
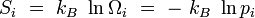
L'entropie totale du système prend alors la forme :
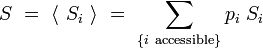

Remplaçons la probabilité par son expression dans le logarithme :
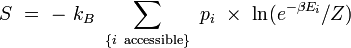
![= \ + \ k_B \ \sum_{\{i \ \mathrm{accessible} \}} \ p_i \ \times \ \left[ \ \beta E_i + \ln Z \ \right]](/pictures/frwiki/51/3d48aac34bfc6f37c0174a4e63a272b9.png)
D'après la définition de la température inverse, on a : kBβ = 1 / T, d'où :
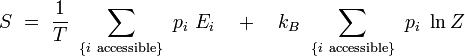
On reconnaît dans le premier terme la valeur moyenne de l'énergie. Par ailleurs, le logarithme de Z est indépendant de l'indice i. On obtient alors en utilisant la condition de normalisation des probabilités :
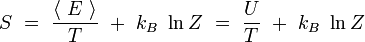
En rapprochant cette formule de celle donnant l'énergie libre F en thermodynamique : F = U - T S, il vient naturellement :
ou encore :
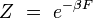
Tableau récapitulatif
Les expressions de F, de U et de S sont suffisantes pour en déduire toutes les autres grandeurs thermodynamiques :
Nom Formule énergie libre de Helmholz 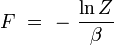
énergie interne 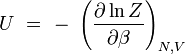
pression 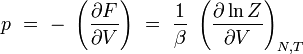
entropie 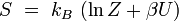
enthalpie libre de Gibbs 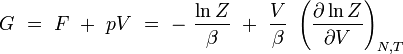
enthalpie 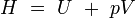
chaleur spécifique à volume constant 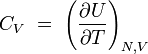
chaleur spécifique à pression constante 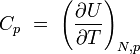
potentiel chimique 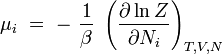
Pour la dernière entrée, il ne s'agit pas de l'ensemble grand-canonique. Il est souvent utile de considérer que l'énergie d'une molécule donnée est distribuée entre plusieurs modes. Par exemple, l'énergie de translation est la partie de l'énergie relative au mouvement du centre de masse de la molécule. L'énergie de configuration se rapporte à la portion de l'énergie associée aux diverses forces attractives et répulsives entre les molécules du système. Les autres modes sont tous considérés comme internes aux molécules. Ils incluent les modes rotationnels, vibrationnels, électroniques et nucléaires. Si nous supposons que chaque mode est indépendant, l'énergie totale peut être exprimée comme la somme de la contribution de chaque composant :
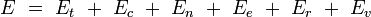
où les indices t, c, n, e, r et v correspondent aux énergies des modes de translation, de configuration, nucléaires, électroniques, rotationnels, et vibrationnels respectivement.
En substituant cette équation dans la toute première équation, nous obtenons :
![Z \ = \ \sum_{ i_j \ \mathrm{niveau \ interne \ a} \ j } \ \left[ \ \prod_{j \, \in \, \{ \, t,c,n,e,r,v \, \}} \ \exp \ ( \, - \, \beta \, E_{ji_j}) \ \right]](/pictures/frwiki/100/d0a1da3db4885b5b94eb94e875f19850.png)
Grace à l'indépendance des modes, on peut permuter la somme et le produit :
![Z \ = \ \prod_{j \, \in \, \{ \, t,c,n,e,r,v \, \}} \ \left[ \ \sum_{ i_j \ \mathrm{niveau \ interne \ a} \ j} \ \exp \ ( \, - \, \beta \, E_{ji_j}) \ \right]](/pictures/frwiki/48/01193aee3d7558fd1552478d71dbbec7.png)
Ainsi, pour chaque mode, on peut définir une fonction de partition associée, et on obtient la fonction de partition totale comme produit de ces fonctions de partition de modes :
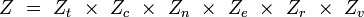
Des expressions simples en sont dérivées pour chacun des modes relatifs à des propriétés moléculaires, telles que les fréquences rotationnelles et vibrationnelles. Les expressions des diverses fonctions de partitions sont données dans la table suivante :
Type Formule nucléaire 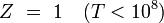
électronique 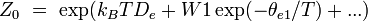
vibrationnel 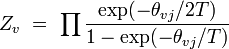
rotationnel(linéaire) 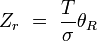
rotationnel (non linéaire) 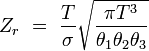
translation 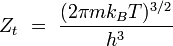
configuration (gaz parfait) 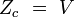
Ces équations peuvent être combinées avec celles de la première table pour déterminer la contribution d'un mode énergetique spécifique aux propriétés thermodynamiques. Par exemple, la « pression de rotation » peut être déterminée de cette manière. La pression totale peut être trouvée en sommant les contributions de pression de tous les modes individuels :
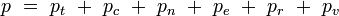
Ensemble grand-canonique
Si le système est ouvert (c'est-à-dire s'il permet l'échange de particules avec l'extérieur), nous devons introduire les potentiels chimiques et remplacer la fonction de partition canonique par la fonction de partition grand-canonique :
![\Xi (V,T,\mu)=\sum_i \exp \left(\beta[\sum_{j=1} ^n \mu_j N_{ij}-E_i]\right)
\,](/pictures/frwiki/56/8c281b28d229ebd89cd8b96ea193521e.png)
où Nij est le nombre de particules de la j-ème espèce dans la i-ème configuration. Il peut arriver aussi que nous ayons d'autres variables à ajouter à la fonction de partition, une variable par quantité conservée. La plupart d'entre elles, cependant, peuvent sans problème être interprétées comme des potentiels chimiques. Dans la plupart des problèmes de matière condensée, les effets sont non relativistes et la masse est conservée. La masse est inversement reliée à la densité, qui est la variable conjuguée de la pression.
Dans le reste de l'article, nous ignorerons cette difficulté et supposerons que les potentiels chimiques ne changent rien. Examinons l'ensemble grand canonique.
Recalculons toutes les expressions en utilisant l'ensemble grand-canonique. Le volume est fixé et ne figure pas dans ce traitement. Comme précédemment, j est l'indice des particules de la j-ème espèce et i est l'indice du i-ème micro-état :
![U=\sum_i {E_i \exp \left(-\beta[E_i-\sum_{j=1} \mu_j N_{ij}]\right)
\over \Xi}\,](/pictures/frwiki/97/a109bc707aa018beeef51730dee3a973.png)
![N_j=\sum_i {N_{ij} \exp \left(-\beta[E_i-\sum_{j=1} \mu_j N_{ij}]\right)
\over \Xi}\,](/pictures/frwiki/97/a6d786f6bfdeb727d45b9a1686eae979.png)
Nom Formule Grand potentiel 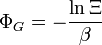
énergie interne 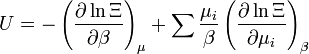
nombre de particules 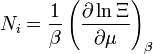
entropie 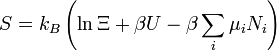
énergie libre de Helmholtz 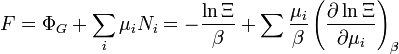
Article annexes
Physique statistique d'équilibre
- Thermodynamique
- Entropie
- Théorie cinétique des gaz
- Statistique de Maxwell-Boltzmann
- Hypothèse ergodique
- Expérience de Fermi-Pasta-Ulam
- Statistique de Fermi-Dirac
- Statistique de Bose-Einstein
- Phénomènes critiques
- Groupe de renormalisation
Physique statistique hors d'équilibre
- Physique statistique hors d'équilibre
- Équation de Boltzmann
- Théorème H
- Hiérarchie BBGKY
- Équation de Vlasov
- Équation de Poisson-Boltzmann
- Expérience de Fermi-Pasta-Ulam
- Théorie ergodique
Bibliothèque virtuelle
- Roger Balian ; Physique fondamentale et énergétique : les multiples visages de l'énergie : cours donné par l'auteur (Service de Physique Théorique du CEA, Saclay) à la première École d'Eté « e2phy » : L'énergie sous toutes ses formes : données scientifiques, contraintes, enjeux, 27-30 août 2001, Site universitaire de Caen.
- Roger Balian ; Entropie, information : un concept protéiforme : texte d'une conférence donnée par l'auteur (Service de Physique Théorique du CEA, Saclay) à l'Université de tous les savoirs (239me conférence : Les États de la matière, 26 août 2000, Conservatoire National des Arts et Métiers, Paris). Publiée par Yves Michaud (éditeur) ; Université de tous les savoirs (Vol. 4), Odile Jacob (2001) pp. 947-959 / Repris en édition de poche : Université de tous les savoirs (Vol. 17), Poches Odile Jacob (2002) pp. 205-220
- Roger Balian ; Le temps macroscopique : texte d'une remarquable conférence sur l'irréversibilité et l'entropie donnée par l'auteur (Service de Physique Théorique du CEA, Saclay) lors du premier colloques "Physique & Interrogations Fondamentales" : Le Temps et sa Flèche organisé par la Société Française de Physique le 8 décembre 1993 à Paris. Publié par : Étienne Klein & Michel Spiro (éditeurs) ; Le Temps et sa Flèche, Les Editions Frontières (1994) pp. 155-211. Repris en poche par Flammarion, Collection Champs (1995).
- Roger Balian ; Entropy, a Protean Concept : texte (en anglais) d'une conférence introductive donnée par l'auteur (Service de Physique Théorique du CEA, Saclay) au séminaire Poincaré du 6 décembre 2003 consacré à l'entropie. Publié dans : J. Dalibard, B. Duplantier et V. Rivasseau (eds.) ; Poincaré seminar 2003, Progress in Mathematical Physics 38, Birkhäuser (2004) 119-144, ISBN .
- [pdf] Cours détaillé
Bibliographie
Vulgarisation
- Bernard Diu, Les atomes existent-ils vraiment ?, Odile Jacob (), ISBN .
- P. Depondt, L’entropie et tout ça - Le roman de la thermodynamique, ISBN .
- Peter W. Atkins, Chaleur & Désordre - Le deuxième Principe de la thermodynamique, Collection "L'univers des sciences", Belin/Pour La Science (1987) 216 pp. Ouvrage de vulgarisation de la thermodynamique des points de vue macroscopique et microscopique. Niveau premier cycle universitaire.
Ouvrages de référence
- Georges Bruhat, Cours de Physique Générale - Thermodynamique, Masson (6e édition-1968) 912 pp. Ce cours de référence, devenu un "classique", est accessible à partir du premier cycle universitaire. Cette 6e édition a été revue et augmentée par Alfred Kastler.
- Yves Rocard, Thermodynamique, Masson (2e édition-1967) 540 pp. Cet autre cours de référence, devenu également un "classique", est accessible à partir du premier cycle universitaire.
Initiation à la physique statistique
- Frederic Reif ; Physique Statistique, Cours de Physique de Berkeley (vol. 5), Armand Colin (1972) 398 pp. réédité par Dunod. Volume 5 du célèbre "Cours de Physique de Berkeley" des années 1960. Accessible à un étudiant du premier cycle universitaire.
- Bernard Jancovici, Thermodynamique & Physique Statistique, Ediscience (1969) 186 pp. Réédité (sans les exercices) par Nathan Université dans sa collection "128 sciences" (1996) 128 pp. Ce petit ouvrage est un cours d'introduction à la thermodynamique via la physique statistique élémentaire. Niveau premier cycle universitaire.
- Percy W. Bridgman, The Nature of Thermodynamics, Harvard University Press (1941) 230 pp. Réflexions sur le sens des 2 principes de la thermodynamique. Ce livre contient quelques équations, accessible au niveau du premier cycle universitaire.
- Mark W. Zemansky & Richard H. Dittman ; Heat & Thermodynamics, McGraw-Hill (6e édition-1981) 544 pp. ISBN 0-07-066647-4. La première moitié de ce volume est un cours de thermodynamique purement macroscopique selon une approche expérimentale : le point de départ est le concept de température usuelle. Cette première partie de l'ouvrage est accessible au niveau du premier cycle universitaire. (La seconde moitié du livre est consacrée à l'approche de la thermodynamique via la physique statistique. Cette partie est plutôt du niveau du second cycle universitaire.)
- Herbert G. Callen, Thermodynamics & an introduction to Thermostatistics, John Wiley & Sons (2e édition-1985) 494 pp. ISBN 0-471-86256-8. Ce livre est le compagnon idéal de l'ouvrage précédent. En effet, la première partie (2/3) de ce volume est un cours de thermodynamique purement macroscopique selon une approche axiomatique : les postulats sont énoncés dès le premier chapitre, le concept de température en est déduit au chapitre suivant. Cette première partie de l'ouvrage est accessible au niveau du premier cycle universitaire, quoique certains développements formels soient d'un niveau plus élevé. (La seconde partie (1/3) du livre est consacrée à l'approche de la thermodynamique via la physique statistique. Cette partie est plutôt du niveau du second cycle universitaire.)
- Ryogo Kubo, Thermodynamics, John Wiley & Sons (1960) pp. Ouvrage classique de thermodynamique. Niveau second cycle universitaire.
- Alfred Brian Pippard, Elements of Classical Thermodynamics - For Advanced Students of Physics, Cambridge University Press (1957) 173 pp. Réédition : avril 2004) ISBN 0-52109-101-2. Niveau second cycle universitaire.
Niveau second cycle universitaire
- (fr) Claudine Guthmann, Danielle Lederer et Bernard Roudet, Éléments de physique statistique, 1996 [détail des éditions].
- (fr) Roger Balian, Du Microscopique au Macroscopique - Cours de Physique Statistique de l'École Polytechnique (2 tomes), Ellipses (1982) 640 pages, (ISBN 2-7298-9000-9) et (ISBN 2-7298-9001-7).
Un cours de physique statistique, qui s'appuie sur la connaissance préalable de la Mécanique Quantique.
- (en) Frederic Reif, Fundamentals of Statistical & Thermal Physics, McGraw-Hill (1965) 651 pages, ISBN 0-07-051800-9.
Ouvrage classique de physique statistique. Niveau second cycle universitaire.
- (en) Linda E. Reichl, A Modern Course in Statistical Physics, John Wiley & Sons (2e édition, 1998) 848 pages, (ISBN 0-471-59520-9).
Un ouvrage moderne déjà classique. Niveau second cycle universitaire.
- (en) Kerson Huang, Statistical Mechanics, John Wiley & Sons (2e édition-1987) 512 pages, (ISBN 0-471-81518-7).
Ouvrage classique de physique statistique. Niveau second cycle universitaire.
- (en) Ryogo Kubo, Statistical Mechanics, John Wiley & Sons (1965) réédité par North-Holland 426 pages, (ISBN 0-444-87103-9).
Ouvrage classique de physique statistique. Niveau second cycle universitaire.
- (en) A.I. Khinchin, Mathematical Foundations of Statistical Mechanics, Dover Publications (1949) 180 pages, (ISBN 0-486-60147-1).
Ouvrage classique sur les fondements de la physique statistique, notamment l'hypothèse ergodique. Niveau second cycle universitaire.
Aspects historiques
- Robert Locqueneux, Préhistoire & Histoire de la thermodynamique Classique (Une histoire de la chaleur), Cahiers d'Histoire & de Philosophie des Sciences n°45, société Française d'Histoire des Sciences & des Techniques (Décembre 1996) 333 pp. ISSN : 0221-3664. Essai sur les théories de la chaleur aux XVIIIe et XIXe siècles. Niveau premier cycle universitaire.
- Jean-Pierre Maury ; Carnot & la machine à vapeur, Collection Philosophies, Presses Universitaires de France (1986) 128 pp. ISBN 2-13-039880-4. Histoire du développement des machines à vapeur depuis leur naissance au XVIIe siècle jusqu'aux travaux théoriques de Carnot ("Réflexions sur la puissance motrice du feu" - 1824) qui posent les fondements de la thermodynamque. Niveau premier cycle universitaire.
- Anouk Barberousse, La Mécanique Statistique - De Clausius à Gibbs, Collection Histoire des Sciences, Belin (2002) 240 pp. ISBN 2-7011-3073-5.Cette collection originale propose une histoire du développement de la théorie cinétique des gaz basée sur des extraits des grands textes fondateurs (traduits en français) mis en perspective contemporaine par une historienne des sciences. Accessible dès le niveau premier cycle universitaire.
- Stephen G. Brush, The Kind of Motion we call Heat - A History of the Kinetic Theories of Gases in the 19th Century (2 vols.), North-Holland (1976). Tome 1 : Physics and the Atomists, ISBN 0-444-87008-3, 300 pages. Tome 2 : Statistical Physics and Irreversible Processes, ISBN 0-444-87009-1, 470 pages. Histoire érudite du développement de la théorie cinétique des gaz, par un professeur de Mécanique des Fluides de l'Université du Maryland (U.S.A.). Après une courte introduction générale (partie A), le premier volume adopte ensuite une approche classée par auteur (partie B). Le second volume (partie C) discute plus spécifiquement certains problèmes, et se termine par une bibliographie (partie D) qui renvoie à la littérature originale. Accessible dès le niveau premier cycle universitaire.
- Peter M. Harman, Energy, Force & Matter - The Conceptual Developpments of 19th Century Physics, Cambridge University Press (1982) pp. ISBN . Histoire du développement de la physique au XIXe siècle. Accessible dès le niveau premier cycle universitaire.
- Peter M. Harman, The Natural Philosophy of James-Clerk Maxwell, Cambridge University Press (1998) 232 pp. ISBN 0-521-00585-X. La philosophie naturelle du professeur Maxwell, fondateur de la théorie de l'électrodynamique et auteur d'importantes contributions en théorie cinétique des gaz. Accessible dès le niveau premier cycle universitaire.
- Carlo Cercignani ; Ludwig Boltzmann - The man who Trusted Atoms, Oxford University Press (1998) 330 pp. ISBN 0-19-850154-4. Biographie scientifique de Ludwig Boltzmann, qui a porté la théorie cinétique des gaz à son acmé. Niveau plutôt second cycle universitaire.
- Paul & Tatiana Ehrenfest ; The Conceptual Foundations of the Statistical Approach in Mechanics, Dover, Inc. (1990) 114 pp. ISBN 0-486-66250-0. Réédition d'un article classique paru initialement en 1912 (en allemand). Niveau second cycle universitaire.
Notes
- ↑ Joseph Willard Gibbs, Principes élémentaires de mécanique statistique, Hermann (1998), ISBN 270566341X. Lire également : Luis Navarro, Gibbs, Einstein and the Foundations of Statistical Mechanics, Archive for History of Exact Sciences 53 147–180 (1998).
- ↑ Domokos Szasz, Botzmann's ergodic hypothesis, a conjecture for centuries ?, Studia Scientiarium Mathematicarum, Hungarica (Budapest) 31 (1996) 299-322.
- ↑ Cf. Frederic Reif ; Physique Statistique, Cours de Physique de Berkeley (vol. 5), Armand Colin (1972), paragraphe 3.5
- ↑ Sont exclus par exemple certains systèmes de spins où l'énergie cinétique des particules serait ignorée. Cf. Reif, op. cit.
- ↑ Cf. Reif, op. cit.
 Portail de la physique
Portail de la physique Portail des probabilités et des statistiques
Portail des probabilités et des statistiques
Catégories : Physique statistique | Thermodynamique
Wikimedia Foundation. 2010.