- Entropie (thermodynamique)
-
Entropie
 Pour les articles homonymes, voir Entropie (homonymie).
Pour les articles homonymes, voir Entropie (homonymie).En thermodynamique, l'entropie est une fonction d'état introduite en 1865[1] par Rudolf Clausius dans le cadre du deuxième principe, d'après les travaux de Sadi Carnot[2]. Clausius a montré que le rapport
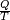 (où Q est la quantité de chaleur échangée par un système à la température T) correspond, en thermodynamique classique, à la variation d'une fonction d’état qu'il a appelée entropie, S et dont l'unité est le joule par kelvin (J/K).
(où Q est la quantité de chaleur échangée par un système à la température T) correspond, en thermodynamique classique, à la variation d'une fonction d’état qu'il a appelée entropie, S et dont l'unité est le joule par kelvin (J/K).La thermodynamique statistique a ensuite fourni un nouvel éclairage à cette grandeur physique abstraite : elle mesure le degré de désordre d'un système au niveau microscopique. Plus l'entropie du système est élevée, moins ses éléments sont ordonnés, liés entre eux, capables de produire des effets mécaniques, et plus grande est la part de l'énergie inutilisée pour l'obtention d'un travail ; c'est-à-dire gaspillée de façon incohérente. Ludwig Boltzmann a exprimé l'entropie statistique en fonction du nombre d’états microscopiques Ω définissant l’état d'équilibre d'un système donné au niveau macroscopique : formule de Boltzmann
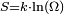 .
.Cette nouvelle définition de l'entropie n'est pas contradictoire avec celle de Clausius. Les deux expressions de l'entropie résultent simplement de deux points de vue différents, selon que l'on considère le système thermodynamique au niveau macroscopique ou au niveau microscopique.
Dans une période récente, le concept d'entropie a été généralisé, et étendu à de nombreux domaines, tels que par exemple :
- l'entropie de Shannon dans le cadre de la théorie de l'information en informatique ;
- l'entropie topologique, ainsi que l'entropie métrique de Kolmogorov-Sinaï, dans le cadre de la théorie des systèmes dynamiques en mathématiques.
Sommaire
L’entropie selon la thermodynamique classique
La non-conservation de l’entropie
La difficulté à donner une définition intuitive de l’entropie d’un système vient du fait qu’elle ne se conserve pas. Elle peut augmenter spontanément lors d’une transformation irréversible. En effet, selon le deuxième principe de la thermodynamique, l’entropie d’un système isolé ne peut pas diminuer, elle augmente ou elle reste constante si la transformation est réversible.
Les transformations réversibles
Une transformation affectant un système thermodynamique est dite réversible si elle est quasistatique et s’effectue sans frottement entraînant un phénomène dissipatif de chaleur. Dans ces conditions, la transformation peut être considérée comme étant constituée d’une succession d’états d’équilibre. Si on inverse le sens de la contrainte du milieu extérieur, responsable de la transformation, on repasse alors par les mêmes états d’équilibre puisqu’il n’y a pas eu de phénomènes dissipatifs. On peut alors modéliser la transformation et décrire parfaitement, à chaque instant, l’état d’équilibre du système.
La transformation réversible est donc un modèle idéal (à rapprocher du modèle idéal du gaz parfait), dont on peut se rapprocher dans les transformations réelles, en s’assurant que la transformation soit très lente, le déséquilibre des variables d'état très faible et en minimisant les frottements.
Remarque : Une transformation réversible qui serait filmée pourrait être projetée à l'envers (c'est-à-dire de la fin au début) sans que la séquence paraisse anormale. C'est par exemple le cas, en première approximation, pour une balle en caoutchouc qui rebondit une fois sur un sol dur, il serait difficile de distinguer si le film est projeté à l'endroit ou à l'envers. En toute rigueur cela est faux car les frottements lors du choc et les frottements de l'air aussi faibles soient-ils rendent le processus irréversible et après plusieurs rebonds, la balle s'arrêterait. Le film à l'envers serait alors choquant puisque la balle rebondirait de plus en plus haut !
À l'inverse, une transformation foncièrement irréversible ne permet pas de faire ce constat, comme dans le cas d'un œuf s'écrasant sur le même sol dur : projeté à l'envers on verrait l'œuf brisé se reconstituer puis monter en l'air. On trouve dans cette interprétation une manifestation de la flèche du temps.
Les transformations irréversibles
Les transformations réelles sont irréversibles à cause de phénomènes dissipatifs. Le système ne peut jamais spontanément revenir en arrière. L’énergie perdue par le système sous forme de chaleur contribue à l’augmentation du désordre global. Or le désordre est mesuré par une fonction d’état appelée entropie : S, introduite par le deuxième principe de la thermodynamique.
Alors que le premier principe est un principe de conservation de l’énergie, le deuxième principe est un principe d’évolution. Il stipule que toute transformation réelle s’effectue avec augmentation du désordre global (système + milieu extérieur) ; le désordre étant mesuré par l’entropie. On dit encore qu’il y a création d’entropie.
L’expression moderne du deuxième principe formalise cette création d’entropie :
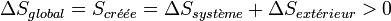
Dans le cas de la transformation idéale réversible, il n’y a pas de création d’entropie :
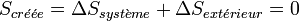
L’entropie comme monnaie d’échange
La thermodynamique classique définit l’entropie comme une grandeur extensive, ce qui signifie que l'on obtient l'entropie d'un système en faisant la somme des entropies de ses parties constituantes (par contraste, la température n’est pas une grandeur extensive parce que la température d’un système n’est pas la somme des températures de ses parties).
Tant que les transformations sont réversibles, on peut raisonner sur l’entropie comme une grandeur conservée. Comme elle peut passer d’un système à un autre ou vers le milieu extérieur, elle est une sorte de monnaie d’échange. Tout système qui gagne de l’entropie la prend à un autre ou au milieu extérieur. L'unité d’entropie, le joule par kelvin (symbole : J·K-1) correspond à la quantité d’entropie gagnée par un système qui reçoit 1 joule de chaleur par kelvin. De façon générale, si un système reçoit
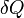 joules de chaleur lors d’une transformation infinitésimale réversible à la température T, alors son entropie augmente de :
joules de chaleur lors d’une transformation infinitésimale réversible à la température T, alors son entropie augmente de :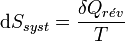 .
.
Cela devient, pour une transformation réversible effectuée à la température T constante :
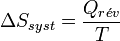 .
.
L’entropie est une fonction d’état. Cela veut dire que sa valeur est déterminée dès que l’état d’équilibre du système est établi. Comme l'entropie d'un corps pur, à la température de zéro kelvin, est nulle car le système est parfaitement ordonné, on peut construire une échelle absolue d'entropie à l'inverse des autres fonctions d'état se déduisant de l'énergie interne qui, elle, n'est pas calculable.
Si la transformation est irréversible, la chaleur mise en jeu devient
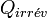 car la chaleur dépend du chemin suivi. De plus, on a vu précédemment qu'il y avait création d'entropie et que le bilan entropique est positif :
car la chaleur dépend du chemin suivi. De plus, on a vu précédemment qu'il y avait création d'entropie et que le bilan entropique est positif :Or la chaleur fournie par le système
est reçue par le milieu extérieur et donc son signe s'inverse 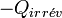 , d'après la règle des signes.
, d'après la règle des signes.La variation de l'entropie du milieu extérieur est alors égale à :
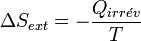 .
.Le bilan entropique devient :
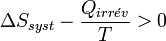
On obtient alors l'inégalité formulée par Clausius pour les transformations irréversibles :
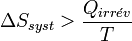
Conséquence de l’inégalité de Clausius sur la quantité de travail fourni par un système
Considérons une transformation effectuée à la température T, qui fait passer un système thermodynamique d’un état initial A à un état final B d’équilibre.
La variation d’entropie du système, associée à cette transformation, peut s'écrire :
On peut procéder de façon réversible ou bien de façon irréversible. Si la variation de la fonction d'état entropie sera la même, il n’en est pas de même pour la chaleur Q et le travail W qui dépendent du chemin suivi et seront donc différents :
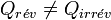 et
et 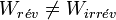 .
.Appliquons le deuxième principe :
d’où
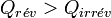 .
.Appliquons le premier principe de conservation de l’énergie interne U :
On déduit que
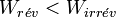 .
.Pour un système (moteur thermique, moteur électrique…) dont la fonction est de fournir du travail au milieu extérieur, le travail fourni est compté négativement d’après la règle des signes : W < 0
Donc ce qui compte c’est la valeur absolue du travail fourni et l’inégalité s’inverse :
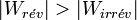
On en déduit que la quantité de travail utile fourni par un système est plus importante lorsque la transformation est réversible.
Remarques :
- Les frottements étant la principale cause d’irréversibilité, on comprend pourquoi on essaye de les minimiser ; c’est le but de la lubrification des pièces en contact et en mouvement dans un ensemble mécanique.
- La vitesse est un facteur d’irréversibilité: la récupération de travail mécanique décroît à mesure que la vitesse d'un véhicule s'accroît. Ainsi, pour une même quantité de carburant, plus un véhicule se déplacera rapidement, plus la distance qu'il pourra parcourir sera réduite.
- Une pile électrique fournit plus de travail électrique si son fonctionnement se rapproche de la réversibilité (faible tension et faible courant de fonctionnement). En revanche si on court-circuite les électrodes, on ne récupère pratiquement que de la chaleur !
Définition de l'entropie selon la physique statistique
La matière est formée de particules (molécules, atomes, électrons...) en perpétuel mouvement (agitation thermique) exerçant les unes sur les autres une force attractive (interactions) dont l'intensité décroit lorsque leur distance mutuelle augmente. Dans un gaz cette distance est relativement grande, les interactions sont donc faibles, de sorte que les particules sont libres de se déplacer dans tout le volume qui leur est offert, mais subissent de nombreuses collisions au cours desquelles leur énergie varie. Dans un liquide les distances mutuelles sont plus petites et les molécules sont moins libres: elles peuvent se déplacer dans tout le volume du liquide mais ne peuvent pas en sortir. Dans un solide chaque molécule est liée élastiquement à ses voisines et vibre autour d'une position moyenne fixe. Il en résulte que, dans tous les cas, la position et l'énergie de chaque particule sont aléatoires.
La somme des énergies de toutes les particules d'un système thermodynamique donné s'appelle l'énergie interne U du système. Lorsque le système est isolé, c'est-à-dire qu'il n'échange ni matière ni énergie avec l'extérieur, on dit que ce système se trouve dans l'ensemble microcanonique. Son état macroscopique est caractérisé par son volume V et son énergie interne U. Cependant les particules peuvent être disposées dans le volume d'un très grand nombre de façons différentes; de même l'énergie interne peut être répartie sur les particules d'un très grand nombre de façons différentes. Chaque façon de placer les molécules dans le volume et de leur distribuer l'énergie interne s'appelle une configuration microscopique de l'état macroscopique caractérisé par le volume V et l'énergie interne U. Le nombre de particules étant, dans un système macroscopique, immensément grand (de l'ordre de 1023) le nombre Ω(U,V) de ces configurations microscopiques est lui-même immensément grand. On définit alors l'entropie S (fonction de U et V) par :
où kB = 1,381.10 − 23J.K − 1 s'appelle la constante de Boltzmann.
Cette relation a été proposée par Ludwig Boltzmann vers les années 1870 alors que la notion d’états microscopiques était encore très spéculative, parce que les atomes et leurs propriétés quantiques étaient mal connus. Il a été en butte à la moquerie des scientifiques de son époque ce qui l'a vraisemblablement conduit au suicide. Aujourd'hui il est considéré comme le père fondateur de la thermodynamique statistique. Sur sa tombe à Vienne est gravée sa formule à l'origine du concept de l'entropie.
Un système évolue en permanence d'une configuration microscopique à une autre, jusqu'à ce qu'il atteigne l'équilibre. On admet alors le principe fondamental suivant :
Étant donné un système isolé en équilibre, il se trouve avec probabilités égales dans chacun de ses micro-états accessibles. Cet axiome est l'énoncé le plus profond du deuxième principe de la thermodynamique.
L'entropie jouit de la propriété d'additivité (on dit que c'est une grandeur extensive) : l'entropie d'un système à l'équilibre est égale à la somme des entropies de chacune de ses parties. Par exemple si l'on sépare le système en deux parties, d'entropies respectives S1 et S2 et ayant pour chacune des parties Ω1 et Ω2 configurations, l'entropie globale S est égale à S1 + S2. D'où :
Il s'ensuit que le nombre de configurations du système Ω = Ω1Ω2.
Équilibre et maximum d'entropie
L'axiome précédent peut être formulé d'une autre manière :
Un système isolé atteint l'équilibre lorsque son entropie devient maximale. Ces deux manières de formuler le principe fondamental de la physique statistique sont équivalentes. La notion d'équilibre est liée à celle de système soumis à des contraintes extérieures. La définition du système est très importante, et on considère ici, que le système se trouve dans l'ensemble microcanonique.
DémonstrationOn peut montrer l'équivalence entre ces postulats à l'aide des multiplicateurs de Lagrange. Dans l'ensemble microcanonique, l'énergie est fixée à une valeur E, et on a donc :
où pi est la distribution de probabilité d'obtenir un résultat i. On cherche alors à maximiser la valeur de l'entropie S avec la contrainte de l'énergie Ei = E fixé, et
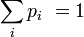 .
.On définit alors la fonctionnelle, dont on cherche le maximum :
Les conditions d'avoir un maximum s'expriment par :
De l'expression 1, on en déduit que la pi est une constante, et avec l'expression 2 (condition de normalisation), on obtient :
On retrouve bien le premier axiome qui affirme que les probabilités associées à chaque micro-états sont équiprobables.
Exemples de compréhension
- Entropie et désordre
Il est courant de dire que l'entropie est une mesure du désordre. En effet, considérons par exemple un jeu de 52 cartes et posons-les toutes du même côté (ordre parfait) ; cet état macroscopique ne peut être réalisé que d'une seule façon : Ω = 1. Retournons 1 carte, ce qui est le début du désordre ; mais il y a Ω = 52 façons de réaliser l'état macroscopique "une seule carte retournée". Le désordre est maximum quand 26 cartes sont d'un côté et 26 cartes de l'autre côté ; le nombre de configurations microscopiques de cet état de désordre maximum est alors Ω = 4,96.1014 ! Dans cet exemple, le nombre de configurations microscopiques (donc l'entropie) est bien une mesure du désordre. Mais il faut être prudent dans l'utilisation de cette notion de désordre, qui est souvent subjective, et lui préférer le nombre Ω de configurations qui est objectif (c'est un nombre).
- Équilibre et maximum d'entropie
Reprenons le jeu de 52 cartes et supposons qu'on les jette en l'air de telle sorte que chaque carte retombe d'un côté ou de l'autre avec la même probabilité. Si l'on recommence l'opération un grand nombre de fois, les valeurs numériques précédentes montrent que le désordre maximum apparaîtra beaucoup plus souvent que toute autre situation.
Considérons maintenant un gaz dans un récipient de volume V. Il comporte non pas 52 molécules mais de l'ordre de 1023. Parmi toutes les façons possibles de ranger ces molécules, il y en a un certain nombre qui laissent la moitié du volume vide (ce qui correspond à toutes les cartes du même côté) mais un nombre immensément plus grand pour lesquelles elles sont uniformément réparties dans tout le volume. Comme toutes ces configurations microscopiques sont équiprobables, la répartition uniforme est réalisée immensément plus souvent que toute autre situation, au point qu'elle apparaît macroscopiquement comme un équilibre stationnaire; et ceci simplement parce que le nombre de configurations microscopiques, et donc l'entropie, qui lui correspondent ont leur valeur maximale. L'équilibre d'un système thermodynamique se produit quand son entropie a la valeur maximale compatible avec les contraintes auxquelles il est soumis (ici la contrainte est le volume).
- Évolution inéluctable vers le désordre
Considérons toujours le jeu de 52 cartes. On les ordonne en les rangeant par ordre décroissant de valeur, de l’as au 2 dans chaque couleur ; les couleurs étant rangées dans l’ordre suivant : trèfle, carreau, cœur et pique. Avec cette contrainte définissant l'ordre parfait, il n’existe qu’une seule configuration ordonnée : Ω(ordonnée) = 1. L’entropie définie selon Boltzmann serait alors égale à :
S = k Ln Ω(ordonnée) = 0 (le système est parfaitement ordonné)
Combien y a-t-il d’arrangements possibles des cartes dans le jeu, c’est-à-dire de configurations ?
Ω = factorielle de 52 = 52 ! = 8.1067
On constate alors que les configurations désordonnées sont extrêmement majoritaires par rapport à la configuration ordonnée.
Supposons maintenant que l’on fasse évoluer le système ordonné en battant le jeu toutes les secondes. Existe-t-il une chance de revenir à l’état initial ordonné ?
Toutes les configurations ont la même probabilité et il y en a 8.1067. Supposons que chaque configuration existe pendant 1 seconde, il faudrait battre le jeu pendant 8.1067 s, soit 2,5.1051 milliards d’années pour avoir peut-être une chance de revenir à l’état ordonné. On peut donc conclure avec certitude, que cette probabilité est quasi-nulle.
Réfléchissons maintenant sur une mole de gaz parfait dans les conditions normales de température et de pression. Le nombre de particules NA = 6,022.1023 est énorme. À l’inverse du jeu précédent où chaque carte est unique et est définie par un seul paramètre, chaque particule de gaz est définie par 3 paramètres de position spatiale et un paramètre d’énergie (agitation thermique). Le nombre de configurations ou complexions est faramineux. Néanmoins, grâce à la thermodynamique statistique, il a été possible de le calculer dans le cas d'un gaz parfait pris dans les conditions normales (volume molaire de 22,4 L) :
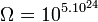 configurations !!!
configurations !!!De plus, il faut remarquer qu’avec l’agitation thermique, le système est en perpétuel changement. Bien évidemment les configurations désordonnées sont les plus nombreuses.
Ce sont ces configurations désordonnées qui occupent la majorité du temps et définissent l’état d’équilibre du système à l’échelle macroscopique.
On pourrait faire la comparaison avec un tableau post-impressioniste de la période pointilliste (voir Seurat, Signac). Quand on s'approche du tableau, on devine tous les différents points de couleur mais quand on s'éloigne suffisamment on a une vision d'ensemble qui est largement influencée par les points de couleur les plus nombreux.
L’application de la formule de Boltzmann permet de ramener la valeur de l'entropie à notre échelle :
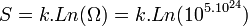
Constante de Boltzmann k = R/NA = 1,381.10-23 J.K-1
S = 159 J.K-1. C’est l’entropie d’une mole de gaz parfait dans les conditions normales.
A zéro Kelvin, l’agitation thermique s’arrête, le gaz se trouve alors dans l'état fondamental de plus basse énergie. Deux cas sont possibles :
- Si l'état fondamental est non-dégénéré, il n’y a plus qu’une seule configuration et l’entropie est nulle S = 0.
- Si l'état fondamental est dégénéré, il existe en général un nombre fini d'états dégénérés. Si g est ce nombre, l'entropie prend sa valeur minimale pour S0 = k ln g.
Remarques d’ordre général
- L'intuition commune comprend déjà difficilement le concept énergie, cette grandeur qui, pour un système isolé, a la propriété de se conserver indéfiniment.
Autrement surprenant est le concept entropie. Pour le même système isolé, l'entropie, dans le meilleur des cas, restera constante, mais en dehors de ce cas très théorique des transformations réversibles, elle ne fera que croître indéfiniment.
Une diminution d'entropie pour un système est néanmoins possible si l’augmentation de l’entropie du milieu extérieur fait plus que compenser la diminution d’entropie du système. Le bilan reste conforme à la deuxième loi de la thermodynamique : une augmentation globale de l'entropie assimilée à une création d'entropie.
Démontrons-le dans le cas d'un système composé d'un vase contenant de l'eau liquide que l'on place à l'air libre à -10 °C soit 263 K. L'eau gèle à 0 °C (273 K) et tant qu'il y a coexistence de glace et d'eau liquide cette température de changement d'état reste constante et égale à 273 K. La chaleur de solidification de l'eau : L(solid) est négative (inverse de la chaleur de fusion qui elle est positive) ainsi que l'entropie de solidification ΔS(syst) = L(solid) / 273 < 0. En revanche, la chaleur est reçue par le milieu extérieur et change de signe (- L(solid)) ; milieu extérieur dont la température n'est pas affectée par l'échange avec le système beaucoup plus petit (notion de source de chaleur). Elle reste constante et égale à 263 K. La variation d'entropie du milieu extérieur est alors égale à :ΔS(ext) = - L(solid) / 263 > 0.
Calculons alors le bilan entropique :
ΔS(syst) + ΔS(ext) = ( L(solid) / 273 ) + ( -L(solid) / 263 ) = L(solid) ( 1/273 - 1/263 ).
Comme L(solid) < 0, il s'ensuit que le bilan est positif et l'entropie créée sera d'autant plus grande que l'écart des températures sera grand ainsi que l'irréversibilité qui va de pair. Si la température du milieu extérieur était très proche de 273 K à -ε près, on se rapprocherait d'une transformation réversible et le bilan entropique serait proche de zéro. En toute rigueur le changement de température entre le système et le milieu extérieur n'est pas brutal. Au voisinage de la paroi séparant le système du milieu extérieur la température varie de façon continue entre 273 K et 263 K. On dit qu'il y a un gradient de température ; phénomène intimement associé à la notion d'irréversibilité.
- L'expression « degré de désordre du système » introduite par Boltzmann peut se révéler ambiguë. En effet on peut aussi définir l'entropie comme une mesure de l'homogénéité du système considéré. L'entropie d'un système thermique est maximale quand la température est identique en tout point. De même, si on verse un liquide colorant dans un verre d'eau, l'entropie du système coloré sera maximale quand, suite au mélange, la couleur du contenu sera devenue uniforme. L'entropie d'un tableau parfaitement lisse et blanc est maximale et ne contient aucune information visible. Si on y ajoute un point coloré, l'entropie diminue, et une information a été ajoutée. Ceci illustre pourquoi, à la naissance de la théorie de l'information, la quantité d'information contenue dans un système était appelée « néguentropie ». Tout système isolé, siège d'une agitation aléatoire, tend spontanément à s'homogénéiser de manière irréversible. C'est pourquoi la notion d'entropie, telle qu’elle est définie par la physique statistique, a été utilisée en théorie de l'information par Claude Shannon au début des années 1950 pour mesurer la perte d'information. voir aussi l'article détaillé : Entropie de Shannon.
- Dans le cadre de sa théorie sur "l'évaporation" des trous noirs, le physicien Stephen Hawking a proposé d'associer une entropie aux trous noirs. En effet, la création et l'annihilation de particules virtuelles à proximité de l'horizon d'un trou noir provoquerait un rayonnement électromagnétique et une perte de masse du trou noir, d'où le terme "évaporation". À ce rayonnement, on associe une température et une entropie. L'évaporation des trous noirs reste à être vérifiée expérimentalement.
Étymologie
Le mot entropie a été inventé par Clausius qui justifie son choix dans Sur diverses formes des équations fondamentales de la théorie mécanique de la chaleur (1865) :
Je préfère emprunter aux langues anciennes les noms des quantités scientifiques importantes, afin qu'ils puissent rester les mêmes dans toutes les langues vivantes ; je proposerai donc d'appeler la quantité S l'entropie du corps, d'après le mot grec η τροπη une transformation. C'est à dessein que j'ai formé ce mot entropie, de manière qu'il se rapproche autant que possible du mot énergie ; car ces deux quantités ont une telle analogie dans leur signification physique qu'une analogie de dénomination m'a paru utile. (cité dans Dictionnaire d'histoire et de philosophie des sciences de Dominique Lecourt, chez PUF, 1999).
Pour en savoir plus
Approche thermodynamique de Carathéodory (1908)
Dès la fin du XIXe siècle, la thermodynamique développée initialement pour les machines thermiques fut appliquée avec succès aux phénomènes électriques et magnétiques, aux changements d'états, et aux réactions chimiques. C'est pourquoi l'introduction traditionnelle de l'entropie basée sur l'étude des machines thermiques cycliques fut critiquée au début du XXe siècle pour son manque de généralité, notamment par Born. Stimulé par cette critique, Carathéodory a remplacé en 1908 cette approche par un traitement purement axiomatique basé sur les propriétés d'intégrabilité des formes différentielles de Pfaff.
En prenant comme postulat de sa thermodynamique d'équilibre qu'il existe des états inaccessibles par voie adiabatique dans le voisinage de tout état d'équilibre donné, Carathéodory[3] démontre l'existence (locale[4]) d'une fonction entropie. En termes techniques, Carathéodory démontre que la forme différentielle « tranfert thermique élémentaire » δQ admet un facteur intégrant 1/T, i.e. que :
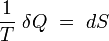
est une différentielle exacte, propriété des fonctions d'état.
Pour voir le lien avec le postulat initial, on remarque que tous les états accessibles par voie adiabatique (δQ = 0) à partir d'un état initial Ei sont alors nécessairement situés sur la surface isentropique S=S(Ei) =cte. Il existe donc des états inaccessibles par voie adiabatique, à savoir tous ceux qui ne sont pas situés sur cette surface. Le théorème de Carathéodory établit la réciproque non-triviale : si il existe des états inaccessibles par voie adiabatique, alors il existe un facteur intégrant, et donc une fonction d'état entropie (Cf. livre de Rocard).
Cette présentation axiomatique reçut à l'époque un accueil enthousiaste de Born, Landé, Chandrasekhar, et Buchdahl, mais son orientation mathématique a rebuté nombre de physiciens, et elle est restée relativement confidentielle jusqu'à ce qu'elle soit simplifiée à la fin des années 1950[5] par L. A. Turner, F. W. Sears, et Peter T. Landsberg[6].
Approche thermodynamique de Lieb-Yngvason (1997)
En 1997, Lieb et Yngvason ont proposé[7] une nouvelle dérivation de l'existence de l'entropie en thermodynamique d'équilibre qui ne fait appel ni aux machines thermiques, ni à la température, ni même aux concepts plus primitifs de « chaud » et de « froid » (et encore moins à la physique statistique). Leur approche est basée sur la notion d'états d'équilibres « accessibles par voie adiabatique » (i.e. dans l'esprit de Carathéodory), mais où leur notion d'accessibilité adiabatique est définie - dans l'esprit de Planck (1926) - de façon purement mécanique à travers le déplacement d'un poids, sans référence au concept de transfert thermique. Dans cette approche, la température apparait à la fin comme une dérivée partielle de l'entropie, une fois que la différentiabilité de cette fonction a été démontrée.
Voir aussi
- Énergie interne
- Énergie libre
- Entropie métrique
- Entropie de Shannon
- Enthalpie
- Enthalpie libre
- Formule de Boltzmann
- Ilya Prigogine
- Stephen Hawking
- Temps
- Théorème H
- Néguentropie
- Principe d'entropie maximale
- Calculs de l'entropie d'un corps pur
Bibliothèque virtuelle
- Roger Balian ; Entropie, information : un concept protéiforme : texte d'une conférence donnée à l'Université de tous les savoirs (239me conférence : Les États de la matière, 26 août 2000, Conservatoire national des Arts et Métiers, Paris). Publiée par Yves Michaud (éditeur) ; Université de tous les savoirs (Vol. 4), Odile Jacob (2001) pp. 947-959 / Repris en édition de poche : Université de tous les savoirs (Vol. 17), Poches Odile Jacob (2002) pp. 205-220
- Roger Balian ; Le temps macroscopique : texte d'une remarquable conférence sur l'irréversibilité et l'entropie donnée lors du premier colloques "Physique & Interrogations Fondamentales" : Le Temps et sa Flèche organisé par la Société Française de Physique le 8 décembre 1993 à Paris. Publié par : Étienne Klein & Michel Spiro (éditeurs) ; Le Temps et sa Flèche, Les Editions Frontières (1994) pp. 155-211. Repris en poche par Flammarion, Collection Champs (1995).
- Olivier Darrigol ; The origins of the entropy concept : texte (en anglais) d'une conférence introductive donnée par l'auteur (REHSEIS-CNRS) au séminaire Poincaré du 6 décembre 2003 consacré à l'entropie. Publié dans : J. Dalibard, B. Duplantier et V. Rivasseau (eds.) ; Poincaré seminar 2003: Bose--Einstein condensation - Entropy, Progress in Mathematical Physics 38, Birkhäuser (2004), ISBN 3-7643-7106-4.
- Roger Balian ; Entropy, a Protean Concept : texte (en anglais) d'une conférence donnée au séminaire Poincaré du 6 décembre 2003 consacré à l'entropie. Publié dans : J. Dalibard, B. Duplantier et V. Rivasseau (eds.) ; Poincaré seminar 2003: Bose--Einstein condensation - Entropy, Progress in Mathematical Physics 38, Birkhäuser (2004), ISBN 3-7643-7106-4.
Indice d'entropie (sciences sociales) : Apparicio & al ; [1]
Bibliographie
Vulgarisation
- W. Atkins ; Chaleur & Désordre - Le Second Principe de la thermodynamique, Collection "L'univers des sciences", Belin/Pour La Science (1987) 216 pp. Par le célèbre professeur de chimie-physique de l'Université d'Oxford, un remarquable ouvrage de vulgarisation de la thermodynamique des points de vue macroscopique et microscopique. Niveau premier cycle universitaire.
- Bernard Diu, Les atomes existent-ils vraiment ?, Odile Jacob (1997), ISBN 2-7381-0421-5.* Peter
Initiation (premier cycle universitaire)
- Bernard Jancovici ; Thermodynamique & Physique Statistique, Ediscience (1969), 186 pp. Réédité (sans les exercices) par Nathan Université dans sa collection "128 sciences" (1996), 128 pp. L'auteur, professeur de physique théorique de l'Université de Paris Sud-Orsay, a longtemps enseigné la physique statistique à l'ENS Ulm (MIP & DEA de physique théorique). Ce petit ouvrage est un cours d'introduction à la thermodynamique via la physique statistique élémentaire. Niveau premier cycle universitaire.
- Frederic Reif ; Physique Statistique, Cours de Physique de Berkeley (vol. 5), Armand Colin (1972), 398 pp. réédité par Dunod. Ce volume 5 du célèbre "Cours de Physique de Berkeley" des années 1960 est un "must" absolu. Sans doute la meilleure introduction à la physique statistique accessible à un étudiant du premier cycle universitaire. Indispensable dans toute bibliothèque de physique digne de ce nom.
- Michel Bertin, Jean-Pierre Faroux & Jacques Renault ; Thermodynamique, Dunod (1976), ISBN 2-04-001703-8.
- Jean-Pierre Faroux & Jacques Renault ; Thermodynamique, Dunod (1997), ISBN 2-10-003099-X.
- Stéphane Olivier & Hubert Gié ; Thermodynamique, Tech & Docs Lavoisier (1996).
- Paul Arnaud ; Cours de chimie physique, Dunod (1990).
- Georges Gonczi ; Comprendre la Thermodynamique, Ellipses (2005). Cours au niveau bac+1 ou bac +2 avec exercices résolus et commentés.
Ouvrages de référence
- Georges Bruhat, Cours de Physique Générale, Thermodynamique, Masson (réimpr. 1968, 6e édition), 912 p..
Ce cours de référence, devenu un « classique », est accessible à partir du premier cycle universitaire. Cette 6e édition a été revue et augmentée par Alfred Kastler, prix Nobel de physique 1966 pour ses travaux en physique atomique, notamment sur le pompage optique, utile au développement des laser. Indispensable dans toute bibliothèque de physique digne de ce nom, cet ouvrage, qui n'est malheureusement plus édité, se trouve d'occasion.
- Yves Rocard, Thermodynamique, Masson (réimpr. 1967 (2e édition)), 540 p..
Père de l'ancien premier ministre Michel Rocard, l'auteur prit, après la Seconde Guerre mondiale, la suite de Georges Bruhat comme directeur du laboratoire de physique de l'École normale supérieure de la rue d'Ulm. Cet autre cours de référence, devenu également un "classique", est accessible à partir du premier cycle universitaire. Indispensable dans toute bibliothèque de physique digne de ce nom, cet ouvrage, qui n'est malheureusement plus édité, se trouve d'occasion.
- Percy W. Bridgman, The Nature of Thermodynamics, Harvard University Press, 1941, 230 p..
Réflexions sur le sens des 2 principes de la thermodynamique. L'auteur, Brigman, a reçu le prix Nobel de physique 1946 pour ses travaux sur les fortes pressions. Ce livre contient quelques équations, accessible au niveau du premier cycle universitaire.
- Mark W. Zemansky & Richard H. Dittman, Heat & Thermodynamics, McGraw-Hill (réimpr. 1981, 6e édition), 544 p. (ISBN 0-07-066647-4).
La première moitié de ce volume est un cours de thermodynamique purement macroscopique selon une approche expérimentale : le point de départ est le concept de température usuelle. Ce livre constitue une mine d'applications. Cette première partie de l'ouvrage est accessible au niveau du premier cycle universitaire. (La seconde moitié du livre est consacrée à l'approche de la thermodynamique via la physique statistique. Cette partie est plutôt du niveau du second cycle universitaire.
- Herbert G. Callen (2e édition-1985) 494 pp., Thermodynamics & an introduction to Thermostatistics, John Wiley & Sons (ISBN 0-471-86256-8).
Ce livre est le compagnon idéal de l'ouvrage précédent. En effet, la première partie (2/3) de ce volume est un cours de thermodynamique purement macroscopique selon une approche axiomatique : les postulats sont énoncés dès le premier chapitre, le concept de température en est déduit au chapitre suivant. Cette première partie de l'ouvrage est accessible au niveau du premier cycle universitaire, quoique certains développements formels soient d'un niveau plus élevé. (La seconde partie (1/3) du livre est consacrée à l'approche de la thermodynamique via la physique statistique. Cette partie est plutôt du niveau du second cycle universitaire.
- A.B. Pippard, Elements of Classical Thermodynamics - For Advanced Students of Physics, Cambridge University Press, 1957) 173 pp. (réimpr. avril 2004) (ISBN 0521091012).
Niveau second cycle universitaire.
- B. Diu, C. Guthmann, D. Lederer, B. Roulet, Physique statistique, Hermann, 1988).
- Roger Balian, Du Microscopique au Macroscopique - Cours de Physique Statistique de l'École Polytechnique, Ellipses, 1982, 2 tomes, 640 p. (ISBN 2-7298-9000-9 , 2-7298-9001-7).
Un beau cours de physique statistique, qui s'appuie sur la connaissance préalable de la Mécanique Quantique. Niveau second cycle universitaire.
- Frederic Reif, Fundamentals of Statistical & Thermal Physics, McGraw-Hill, 1965, 651 p. (ISBN 0-07-051800-9).
Ouvrage classique de physique statistique. Niveau second cycle universitaire.
- Linda E. Reichl, A Modern Course in Statistical Physics, John Wiley & Sons (réimpr. 1998, 2e édition), 848 p. (ISBN 0-471-59520-9).
Un ouvrage moderne déjà classique. Linda Reichl est professeur de physique statistique à l'Université d'Austin, Texas (USA). Niveau second cycle universitaire.
- Kerson Huang (2e édition-1987) 512 pp., Statistical Mechanics, John Wiley & Sons (ISBN 0-471-81518-7).
Ouvrage classique de physique statistique. Niveau second cycle universitaire.
- Ryogo Kubo, Thermodynamics, John Wiley & Sons, 1960.
Ouvrage classique de thermodynamique. Niveau second cycle universitaire
- Ryogo Kubo, Statistical Mechanics, John Wiley & Sons, 1965 (réimpr. par North-Holland), 426 p. (ISBN 0-444-87103-9).
Ouvrage classique de physique statistique. Niveau second cycle universitaire.
- Andreas Greven, Gerhard Keller, & Gerald Warnecke (éditeurs), Entropy, Princeton University Press, 2003 (ISBN 978-0-691-11338-8)
Aspects historiques
- Jean-Pierre Maury ; Carnot & la machine à vapeur, Collection Philosophies, Presses Universitaires de France (1986) 128 pp. ISBN : 2-13-039880-4. Histoire du développement des machines à vapeur depuis leur naissance au XVIIe siècle jusqu'aux travaux théoriques de Carnot (Réflexions sur la puissance motrice du feu - 1824) qui posent les fondements de la thermodynamique. Niveau premier cycle universitaire.
- Anouk Barberousse ; La Mécanique statistique - De Clausius à Gibbs, Collection Histoire des Sciences, Belin (2002) 240 pp. ISBN : 2-7011-3073-5.Cette collection originale propose une histoire du développement de la théorie cinétique des gaz basée sur des extraits des grands textes fondateurs (traduits en français) mis en perspective contemporaine par une historienne des sciences (CNRS). Accessible dès le niveau premier cycle universitaire.
- Stephen G. Brush ; The Kind of Motion we call Heat - A History of the Kinetic Theories of Gases in the 19th Century (2 vols.), North-Holland (1976). Tome 1 : Physics and the Atomists, ISBN : 0-444-87008-3, 300 pages. Tome 2 : Statistical Physics and Irreversible Processes, ISBN : 0-444-87009-1, 470 pages. Histoire érudite du développement de la théorie cinétique des gaz, par un professeur de Mécanique des Fluides de l'Université du Maryland (USA). Après une courte introduction générale (partie A), le premier volume adopte ensuite une approche classée par auteur (partie B). Le second volume (partie C) discute plus spécifiquement certains problèmes, et se termine par une bibliographie (partie D) qui renvoie à la littérature originale. Accessible dès le niveau premier cycle universitaire.
- Peter M. Harman ; Energy, Force & Matter - The Conceptual Developpments of 19th Century Physics, Cambridge University Press (1982) pp. ISBN. Histoire du développement de la physique au XIXe siècle par un professeur d'Histoire des Sciences de l'Université de Lancaster (UK). Accessible dès le niveau premier cycle universitaire.
- Peter M. Harman ; The Natural Philosophy of James-Clerk Maxwell, Cambridge University Press (1998) 232 pp. ISBN : 0-521-00585-X. La philosophie naturelle du génial professeur Maxwell, fondateur de la théorie de l'électrodynamique et auteur d'importantes contributions en théorie cinétique des gaz, par un professeur d'Histoire des Sciences de l'Université de Lancaster (UK) responsable de l'édition des œuvres scientifiques du Maître. Accessible dès le niveau premier cycle universitaire.
- Carlo Cercignani ; Ludwig Boltzmann - The man who Trusted Atoms, Oxford University Press (1998) 330 pp. ISBN : 0-19-850154-4. Biographie scientifique du grand professeur Boltzmann, qui a porté la théorie cinétique des gaz à son acmé. Par un professeur de physique mathématique de l'Université de Milan (Italie), spécialiste de l'"équation de Boltzmann". Niveau plutôt second cycle universitaire.
- Paul & Tatiana Ehrenfest ; The Conceptual Foundations of the Statistical Approach in Mechanics, Dover, Inc. (1990) 114 pp. ISBN : 0-486-66250-0. Réédition d'un article classique paru initialement en 1912 (en allemand). Niveau second cycle universitaire.
Notes
- ↑ chapitre 1 du Lev Landau et Evguéni Lifchitz, Physique théorique, tome 5 : Physique statistique, éd. MIR, Moscou [détail des éditions]
- ↑ Sadi Carnot ; Réflexions sur la puissance motrice du feu et sur les machines propres à développer cette puissance, (1824). Réédition Jacques Gabay (2005), ISBN 2-87647-069-1.
- ↑ Constantin Carathéodory, Untersuchungen über die Grundlagen der Thermodynamik, Mathematische Annalen 67 (1909), 355-86, reproduit dans : Gesammelte Mathematische Schriften (Munich, 1955), pp. 131-66.
- ↑ Sur ce point , lire e.g. : Jos Uffink ; Bluff your way in the second law of thermodynamics, Studies in History and Philosophy of Modern Physics (2000). ArXiv : cond-mat/0005327.
- ↑ On trouve cette approche simplifiée exposée par exemple dans : Peter T. Landsberg ; Thermodynamics, John Wiley & Sons (1961), et : I. P. Bazarov, Thermodynamics, Oxford University Press (1964).
- ↑ Peter T Landsberg ; On suggested simplifications of Caratheodory’s thermodynamics, Phys.Status Solidi 1 (1961), 120 ; Peter T Landsberg ; A deduction of Caratheodory’s principle from Kelvin’s principle, Nature 201 (1964), 485 ; Peter T Landsberg ; The Born centenary: Remarks about classical thermodynamics, Am J Phys 51 (1983), 842. H.Meyer-Ortmanns, Peter T Landsberg, S Abe, A.K.Rajagopal & T.Yamano ; A note on the limitations of standard thermodynamics, Annalen der Physik 11 (2002), 457.
- ↑ Elliott H. Lieb & Jakob Yngvason ; A fresh look at entropy and the Second Law of Thermodynamics, Physics Today 53 (2000), 32-37. ArXiv : math-ph/0003028. Elliott H. Lieb & Jakob Yngvason ; The Physics and Mathematics of the Second Law of Thermodynamics, Physics Report 310 (1999), 1-96. ArXiv : cond-mat/9708200. Elliott H. Lieb & Jakob Yngvason ; A Guide to Entropy and the Second Law of Thermodynamics, Notices of the American Mathematical Society 45 (1998), 571-581. ArXiv : math-ph/9805005.
 Portail de la physique
Portail de la physique Portail de la chimie
Portail de la chimie
Catégories : Thermodynamique | Thermochimie | Physique mathématique | Statistiques | Théorie de l'information | Grandeur physique | Système
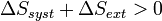
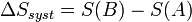
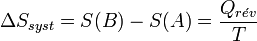
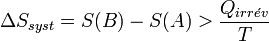
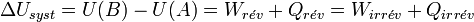
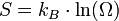
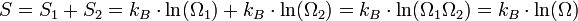

![S_M \left(p_i, \lambda\right) = S + \lambda \left[ \sum_i p_i -1 \right]](/pictures/frwiki/56/842b63c67518049d654843c4597660e3.png)
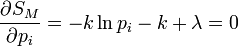
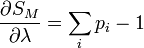
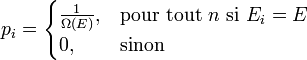
Wikimedia Foundation. 2010.