- Contrôle thermodynamique
-
Contrôle cinétique et contrôle thermodynamique
En chimie, quand une réaction chimique peut mener à plus d'un produit, la sélectivité peut parfois dépendre des conditions de réaction et peut même être inversée. Le contrôle cinétique se dit des conditions de réaction qui favorisent surtout le produit formé le plus rapidement, alors que le contrôle thermodynamique se dit des conditions qui donnent surtout le produit le plus stable. Le contraste entre ces deux régimes n'est utile que si les deux types de conditions favorisent différents produits, ou, dans les cas contraires, si la sélectivité pour le produit dominant soit plus favorable sous un régime que sous l'autre.
Sommaire
Exemples
- La déprotonation d'une cétone asymétrique.
- Lors de la déprotonation d'une cétone asymétrique, le produit cinétique est l'énolate résultant du retrait du α-H le plus accessible alors que le produit thermodynamique possède un groupement énolate plus substitué. L'utilisation de températures basses et de bases encombrées augmente la sélectivité cinétique. La différence en pKb de la base et de l'énolate est tellement grande que la réaction est à toutes fins pratiques irréversible. Donc, l'équilibration menant au produit thermodynamique est probablement un échange de proton pendant l'addition entre l'énolate cinétique et la cétone encore intouchée. Une addition inverse (ajout de la cétone à la base) avec mélange rapide amoindrirait la probabilité de cet échange. La position de l'équilibrium dépendra du cation et du solvant choisis.
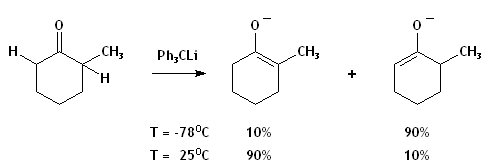
- Si une base beaucoup moins forte est utilisée, la déprotonation sera incomplète, et il y aura équilibre entre réactifs et produits.[1] Un contrôle thermodynamique s'imposera, mais la réaction restera incomplète à moins de piéger l'énolate, comme dans l'exemple ci-dessous. Puisque les transferts de H sont très rapides, la réaction-piège étant plus lente, le rapport des produits piégés miroitera largement l'équilibre gouvernant la déprotonation.
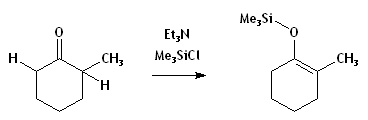
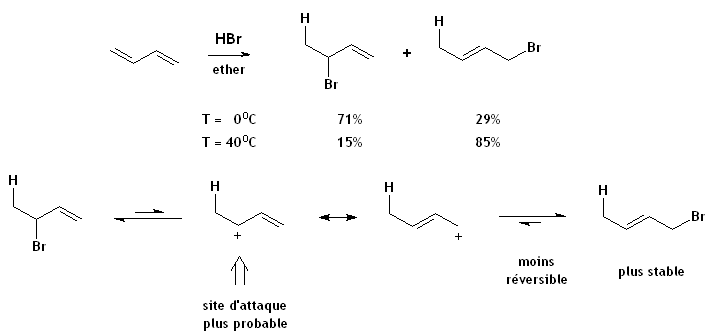
- L'addition de HBr sur un diène.
- La réaction d'addition du HBr sur le buta-1,3-diène à températures élevées donne surtout le 1-bromo-2-butène, le produit le plus stable résultant de l'addition 1,4, mais à températures basses donne plutôt le 3-bromo-1-butène, le produit cinétique d'addition 1,2. L'explication des ces sélectivités inversées est comme suit: Les deux réactions débutent avec la protonation selon Markovnikov à la position 1, donnant le cation allylique stabilisé par mésomérie. L'addition 1,4 place le plus gros atome Br sur un carbone moins congestionné et donne une groupement alcène plus substitué, tandis que l'addition 1,2 procède par attaque par le nucléophile (Br-) sur le carbone du cation allylique le plus apte à soutenir la charge positive (parce que plus substitué).
Caractéristiques
- Cette question de contrôle cinétique ou thermodynamique ne se pose que si le chemin réactionnel se coupe. C'est à dire si le produit cinétique (moins stable donc plus haut en énergie) passe par un état de transition plus faible (donc moins haut en énergie).
- Toute réaction mène d'abord au produit formé le plus aisément et est donc a priori sous contrôle cinétique au départ.[2]
- Le contrôle thermodynamique nécessite un mécanisme d'équilibration. Pour qu'il y ait contrôle thermodynamique, il faut que soit possible un produit plus stable (qui est toutefois formé moins rapidement), que le résultat initial soit réversible (ou qu'il existe un mécanisme d'échange entre les deux produits), et qu'il y ait suffisamment de temps écoulé pour que le produit plus stable accumule. La formation du produit plus stable sera nécessairement moins réversible et celui-ci viendra à dominer sur l'autre jusqu'à ce que l'équilibre entre les deux soit enfin établi.
- Sous un régime purement cinétique, la distribution des produits dépendra uniquement des vitesses de formation des deux produits, lesquelles vitesses dépendent des énergies d'activation Ea ou ΔG‡. En tout temps t, le rapport des concentrations des deux produits P1 et P2 sera égal au rapport des constantes de vitesses k1 et k2 et donc fonction de la différence entre les énergies d'activation,
![ln(\frac {[P_1]_t}{[P_2]_t}) = ln(\frac {k_1}{k_2}) = -\frac {\Delta E_a}{RT}](/pictures/frwiki/56/8d1ec35588480a43b8fb705936d9f473.png) (équation 1)
(équation 1)
- À moins que l'équilibration soit empêchée, le contrôle purement cinétique est pratiquement impossible, puisque l'équilibration s'entame avant même que les réactifs n'aient été complètement consommés.
- Sous un régime purement thermodynamique, l'équilibre aura été atteint et la distribution des produits sera uniquement fonction des stabilités G°. Après un temps infini, le rapport des concentrations des deux produits sera égal à la constante d'équilibre Keq et donc fonction de la différence entre les énergies de Gibbs,
![ln(\frac {[P_1]_{\infty}}{[P_2]_{\infty}}) = ln\ K_{eq} = -\frac {\Delta G^\circ}{RT}](/pictures/frwiki/101/e3163b888477e831bb5179f79ad2b9a8.png) (équation 2)
(équation 2)
- En général, les temps de réaction relativement courts favoriseront le contrôle cinétique, alors que les réactions prolongées favoriseront le contrôle thermodynamique. Les températures plus basses augmenteront la sélectivité sous les deux régimes, puisque T paraît dans le dénominateur dans les deux cas. La température idéale pour optimiser le rendement du produit favorisé par la cinétique sera la température la plus basse qui permettra toutefois l'achèvement de la réaction dans un temps raisonnable.[3] Au contraire, les températures élevées permettront que l'équilibre s'établisse dans un temps raisonnable pour favoriser le produit le plus stable, mais diminuera la sélectivité;[4] après que l'équilibre aura été atteint, on peut refroidir lentement pour déplacer l'équilibre davantage vers le produit le plus stable.
- Si une réaction est sous contrôle thermodynamique à une température donnée, elle le sera aussi à une température supérieure après le même temps de réaction.
- En contrepartie, si une réaction est sous contrôle cinétique à une température donnée, elle le sera aussi à une température inférieure après le même temps de réaction.
- En présumant qu'une nouvelle réaction est a priori sous contrôle cinétique, on décèle l'opération d'un mécanisme d'équilibration (et donc la possibilité d'un contrôle thermodynamique éventuel) si la distribution des produits:
- évolue avec le temps,
- indique la dominance d'un produit à une température mais la dominance d'un autre à une température différente (inversion de la dominance), ou
- change avec la température mais ne s'explique pas par l'équation 1 ci-haut, c'est à dire qu'une augmentation de la température (sans changer le temps de réaction) occasionne un changement dans le rapport des deux produits [A]t / [B]t qui est plus petit ou plus grand qu'anticipé par le simple changement de la température, pourvu que ΔEa soit largement indépendant de la température sur une plage de température modeste.[5]
- De même manière, on décèle la possibilité d'un contrôle cinétique éventuel si une réduction de la température occasionne un changement dans le rapport des deux produits qui ne s'explique pas par l'équation 2 ci-haut, pourvu que
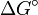 soit largement indépendant de la température sur une plage de température modeste.[6]
soit largement indépendant de la température sur une plage de température modeste.[6]
Notes et références
- ↑ La déprotonation sera quand même plus rapide au site le moins encombré.
- ↑ Sauf si l'équilibration qui suit est aussi rapide ou plus rapide.
- ↑ À moins de se contenter d'une réaction inachevée, ce qui peut nécessiter une séparation du réactif inutilisé du produit désiré.
- ↑ Au pire, le produit constituera 50% du mélange réactionnel.
- ↑ ΔEa sera indépendante de la température, ou presque, si
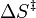 est petit, ce qui sera le cas si l'étape déterminante de la vitesse menant à chacun des produits est de la même molécularité, par exemple si ces deux procèdent par une collision avec le même réactif.
est petit, ce qui sera le cas si l'étape déterminante de la vitesse menant à chacun des produits est de la même molécularité, par exemple si ces deux procèdent par une collision avec le même réactif. - ↑ sera indépendante de la température, ou presque, si
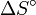 est petit, ce qui sera le cas si la transformation globale donnant un des produits est de la même molécularité que celle donnant l'autre, par exemple si ces deux sont des fragmentations d'une molécule en deux, ou une condensation de deux molécules en une.
est petit, ce qui sera le cas si la transformation globale donnant un des produits est de la même molécularité que celle donnant l'autre, par exemple si ces deux sont des fragmentations d'une molécule en deux, ou une condensation de deux molécules en une.
Voir aussi
Bibliographie
- (en) Organic Chemistry, 3rd ed., M. A. Foxe & J. K. Whitesell, Jones & Bartlett, 2004 ISBN 0-7637-2197-2
- (en) A Guidebook to Mechanism in Organic Chemistry, 6th Edition, Peter Sykes, Pearson Prentice Hall, 1986. ISBN 0-582-44695-3
- (en) Introduction to Organic Chemistry I, Seth Robert Elsheimer, Blackwell Publishing, 2000 ISBN 0632044179
 Portail de la chimie
Portail de la chimie
Catégorie : Génie chimique
Wikimedia Foundation. 2010.