- Polyèdre de coordination
-
La notion de polyèdre de coordination est utilisée en cristallographie et en chimie pour décrire l'environnement d'une espèce chimique par ses ligands dans un cristal ou un complexe. Il s'agit du polyèdre formé par les ligands autour de l'espèce chimique centrale. La coordinence, c'est-à-dire le nombre de ligands, n'est pas suffisante pour décrire l'environnement d'une espèce chimique, car pour une coordinence donnée, il peut exister plusieurs polyèdres de coordination. Par exemple, à l'état solide, le cuivre a une coordinence 4 d'oxygène dans le silicate de baryum et de cuivre BaCuSi2O6[1] et également une coordinence 4 de soufre dans la stannite Cu2FeSnS4[2], mais son polyèdre de coordination est un carré CuO4 dans BaCuSi2O6 et un tétraèdre CuS4 dans la stannite.
En solution la notion de coordinence est souvent réduite à la coordinence des cations (les anions sont coordonnés avec une énergie de liaison faible qui ne donne pas une bonne stabilité à la structure). La coordinence est définie, comme à l'état solide, par l'environnement du cation dans sa première sphère de coordination. Celle-ci est définie par les ligands (éventuellement le solvant) qui sont directement liés au cation. Ces ligands sont géométriquement orientés, et leur liaison est raisonnablement forte pour que les échanges avec les entités de la seconde sphère de coordination soient lents. Cette seconde sphère est surtout constituée de molécules de solvant, mais pas exclusivement, qui sont orientées par la présence du cation, mais qui s'échangent (1) rapidement avec les molécules de solvant qui lui sont extérieures, et (2) lentement avec les ligands qui lui sont intérieurs. La géométrie de la coordinence d'un cation en solution est moins variée qu'à l'état solide. Avec les ligands courants, la plupart des cations métalliques adoptent une coordinence octaédrique, une faible part adopte une coordinence tétraédrique ou plan carré, et les métaux de la colonne du cuivre au degré d'oxydation I ont une coordinence deux (linéaire). De ces derniers, l'ion diammine argent Ag(NH3)2+ (appelé aussi réactif de Tollens) est le plus connu. Les coordinences autres résultent de la présence de ligands particuliers (par exemple encombrants), ou de cations métalliques de grosse taille (Ta, W, etc.)
Les polyèdres de coordination possibles pour une espèce chimique donnée et ses ligands dépendent du degré d'oxydation de l'espèce chimique et des rayons ioniques mis en jeu, plus précisément du rapport rc / ra où rc est le rayon ionique de l'espèce chimique centrale et ra le rayon de ses ligands.
Les polyèdres de coordination ne sont en général pas réguliers. Par exemple, dans le cas des complexes, ils peuvent présenter une déformation due à la configuration électronique du cation métallique dans le champ cristallin créé par les ligands. C'est le cas de Cu(H2O)62+ qui présente une distorsion axiale qui s'explique par l'Effet Jahn-Teller. Un autre cas de non régularité est celui de la nature chimiques différentes des ligands, comme avec Cu(NH3)4(H2O)22+. Dans tous les cas, il est cependant d'usage d'utiliser le nom des polyèdres réguliers pour décrire la coordination des espèces chimiques.
Sommaire
Les différents polyèdres de coordination
Les polyèdres de coordination sont généralement constitués de faces triangulaires équilatérales, comme le tétraèdre, l'octaèdre, la bipyramide trigonale ou l'icosaèdre. On rencontre aussi souvent des faces carrées, comme dans la pyramide tétragonale, le cube ou le cuboctaèdre. Il est plus rare de trouver des polyèdres de coordination dont les faces possèdent un grand nombre de sommets.
Le tableau suivant donne les polyèdres de coordination rencontrés dans les complexes en solution et dans les cristaux[3] :
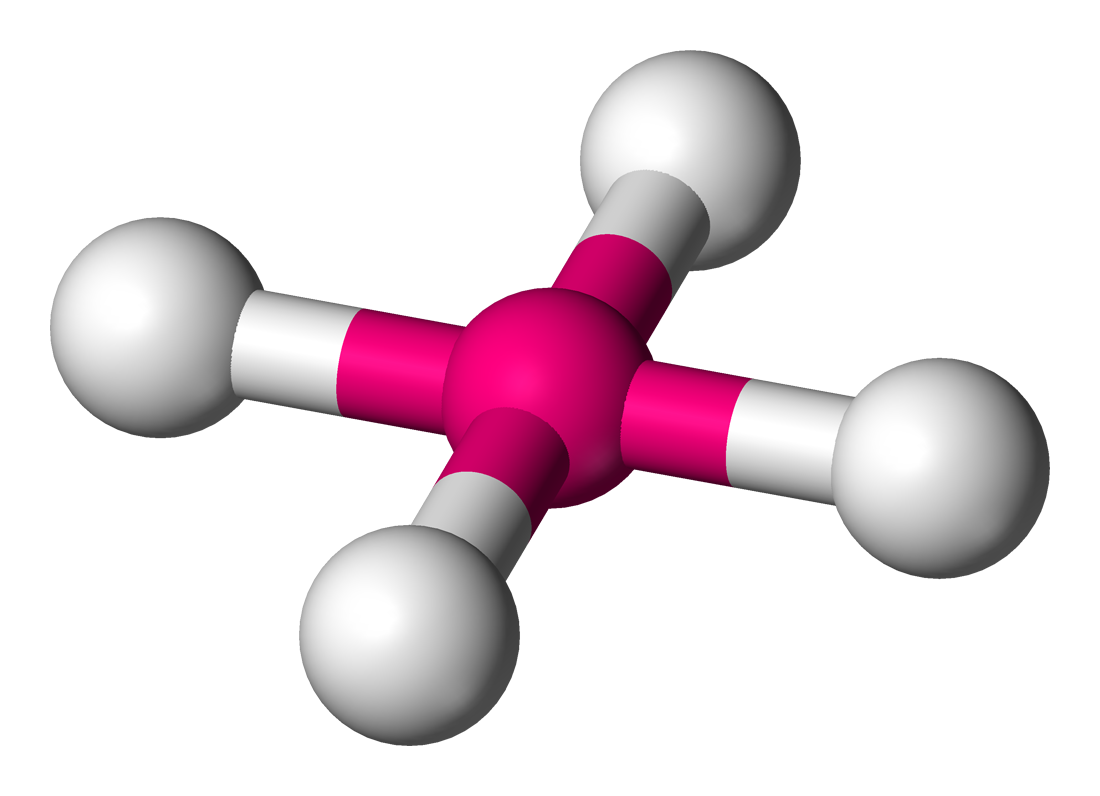
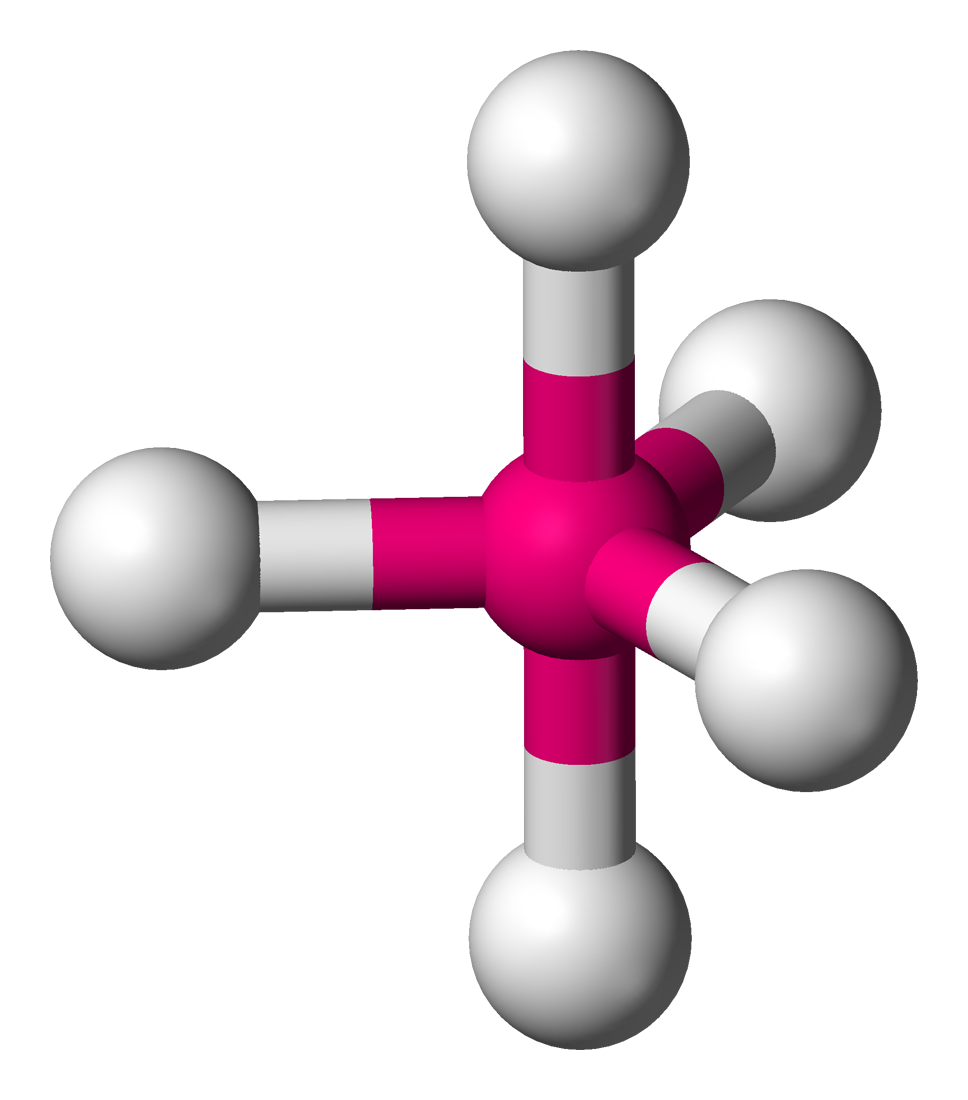
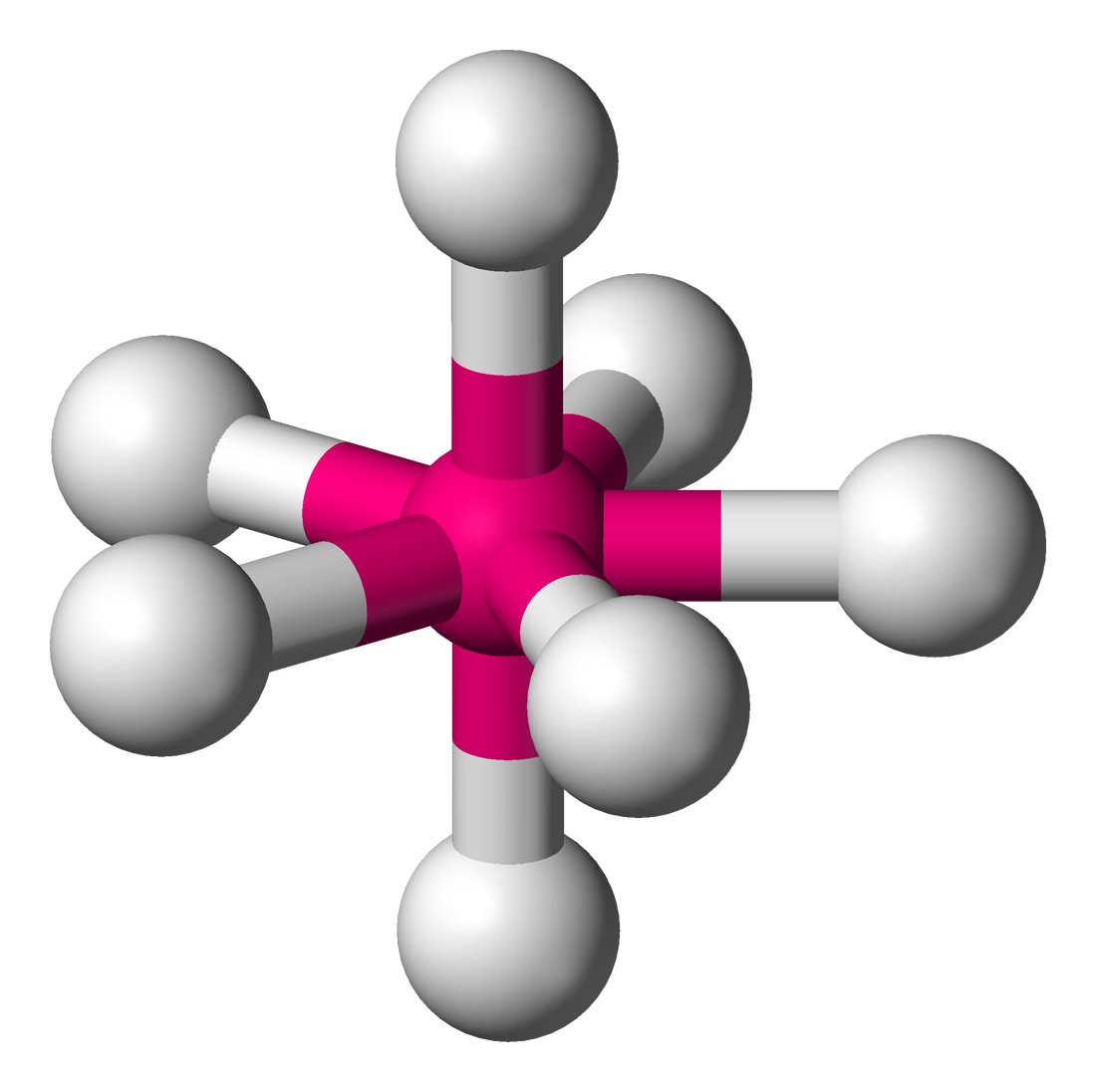
Coordinence Polyèdre Représentation Exemples : structure moléculaire Exemples : état solide 2 linéaire 
Cu dans l'ion diméthylcuprate(I) (CH3)2Cu- Ag dans le dicyanoargentate(I) de potassium
KAg(CN)2[4]3 triangle 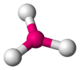
B dans le trifluorure de bore BF3 NO3 dans le salpêtre KNO3[5],
BO3, CO3 dans la calcite CaCO3[6]4 tétraèdre 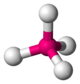
Ni dans le tétracarbonylnickel(0)[7] Ni(CO)4 SiO4 dans le quartz SiO2[8], BO4 carré 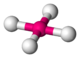
Ni dans l'ion tétracyanonickelate(II)[9] Ni(CN)42- Cu dans CuO[10] 5 bipyramide
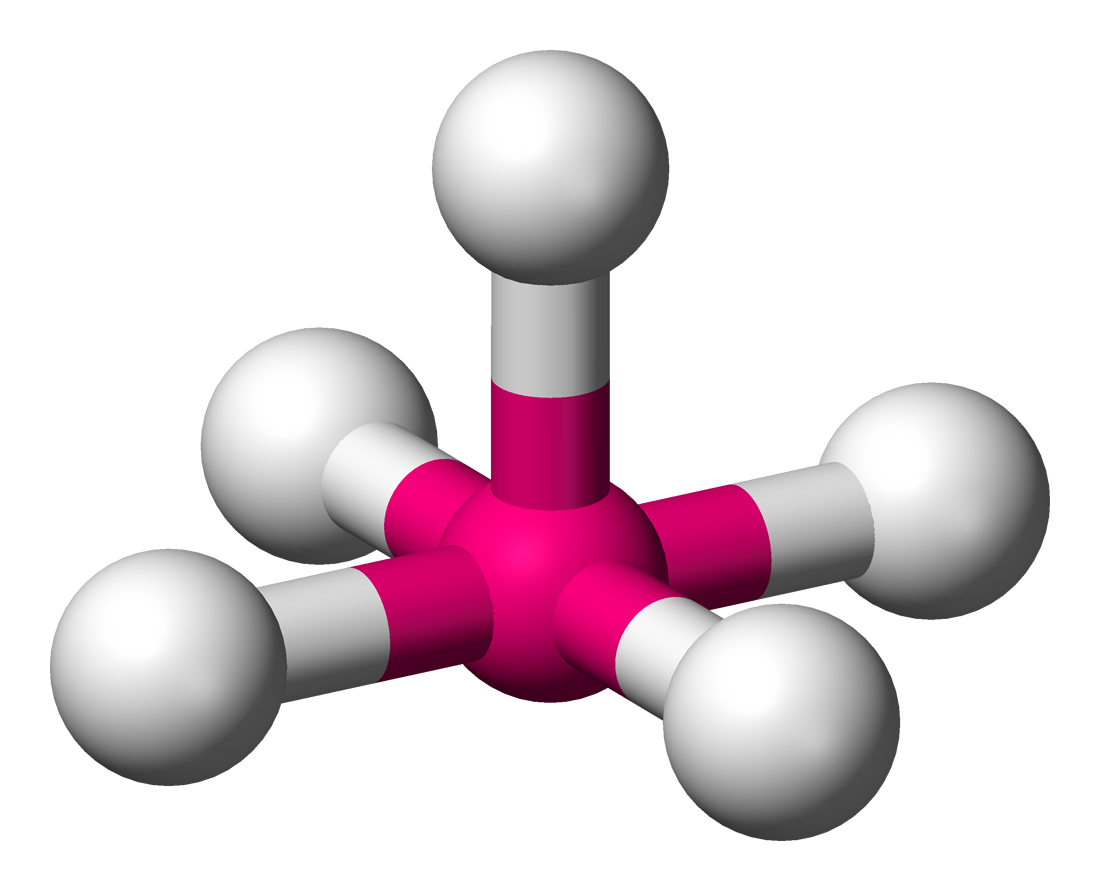
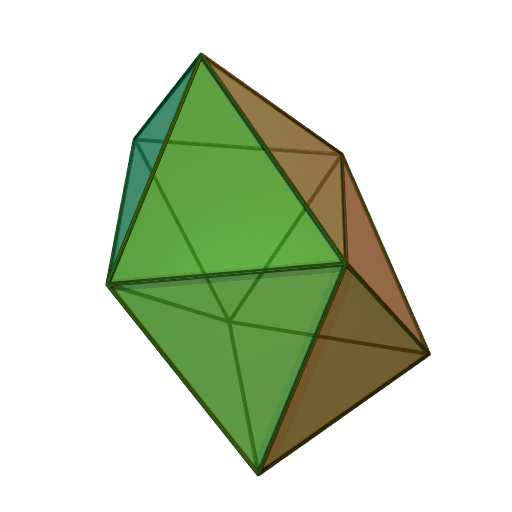
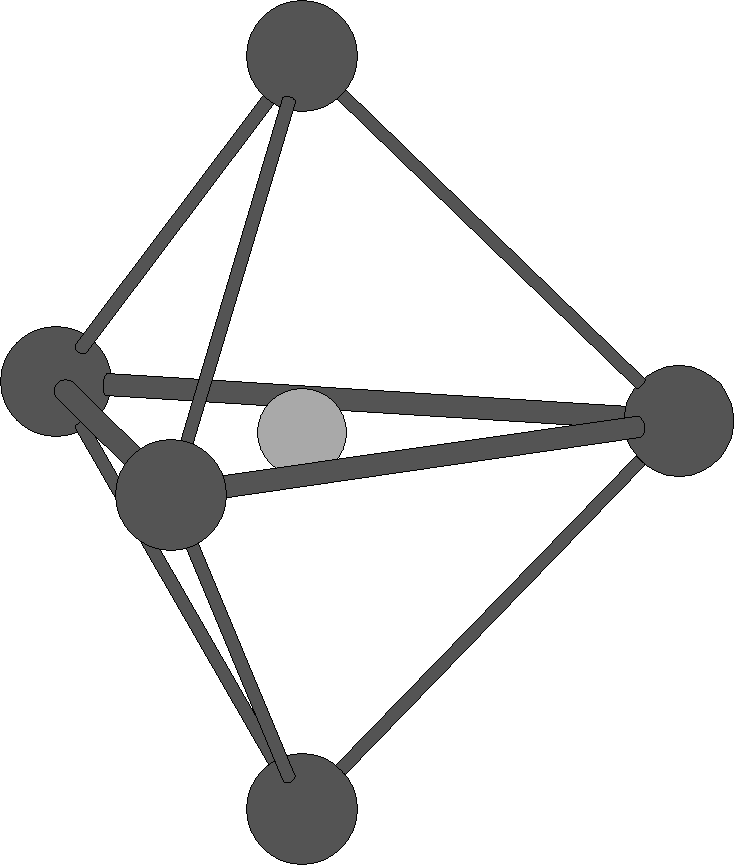
trigonale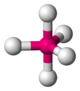
Fe dans le pentacarbonylfer(0) Fe(CO)5 K dans K3Sb[11] pyramide
tétragonale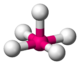
Pas de structure courante simple, mais c'est une structure classique intermédiaire dans une substitution sur un complexe plan-carré (par addition du ligand entrant) ou pour dans une substitution sur un complexe octaédrique (après le départ du ligand sortant)[12] V dans CaV4O9[13],
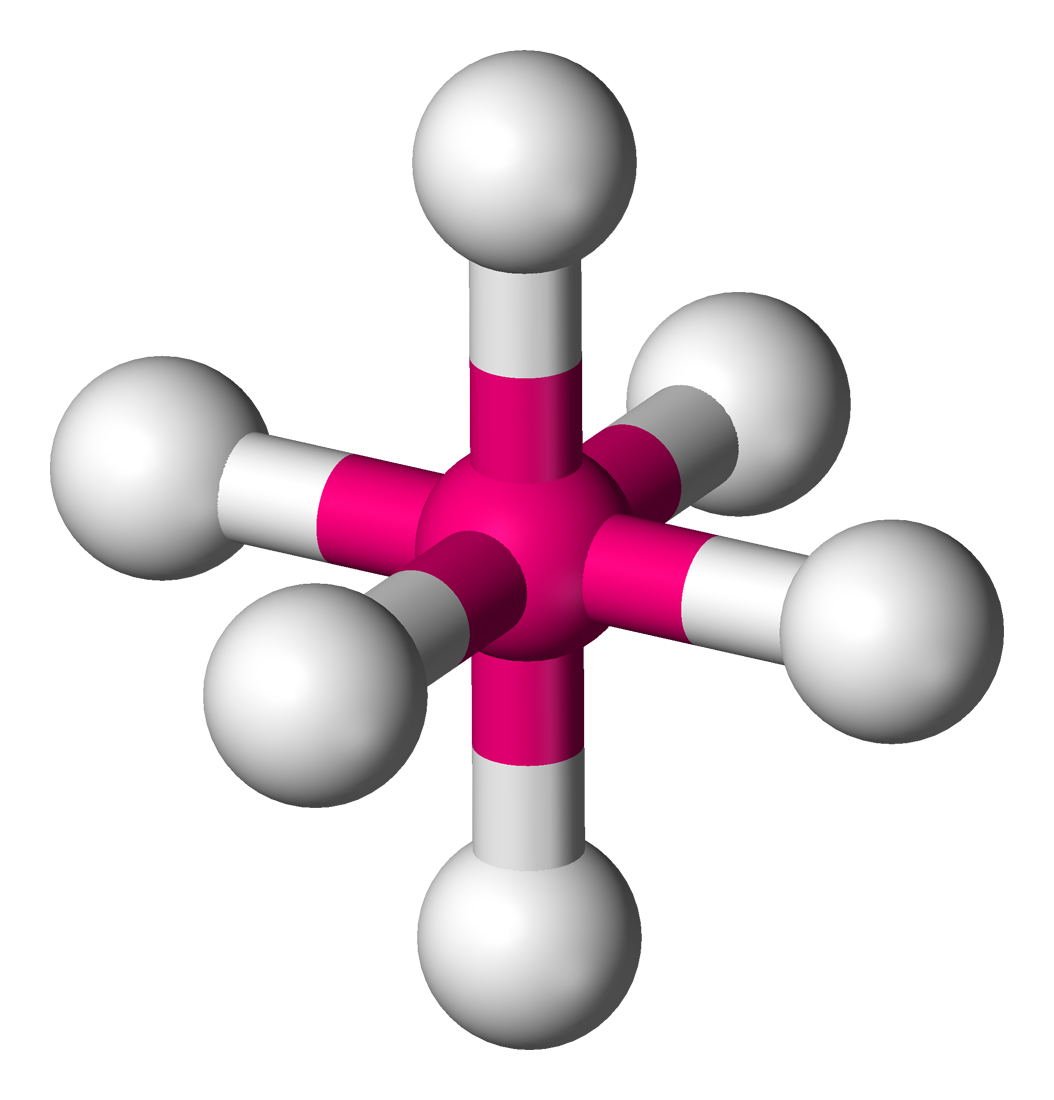
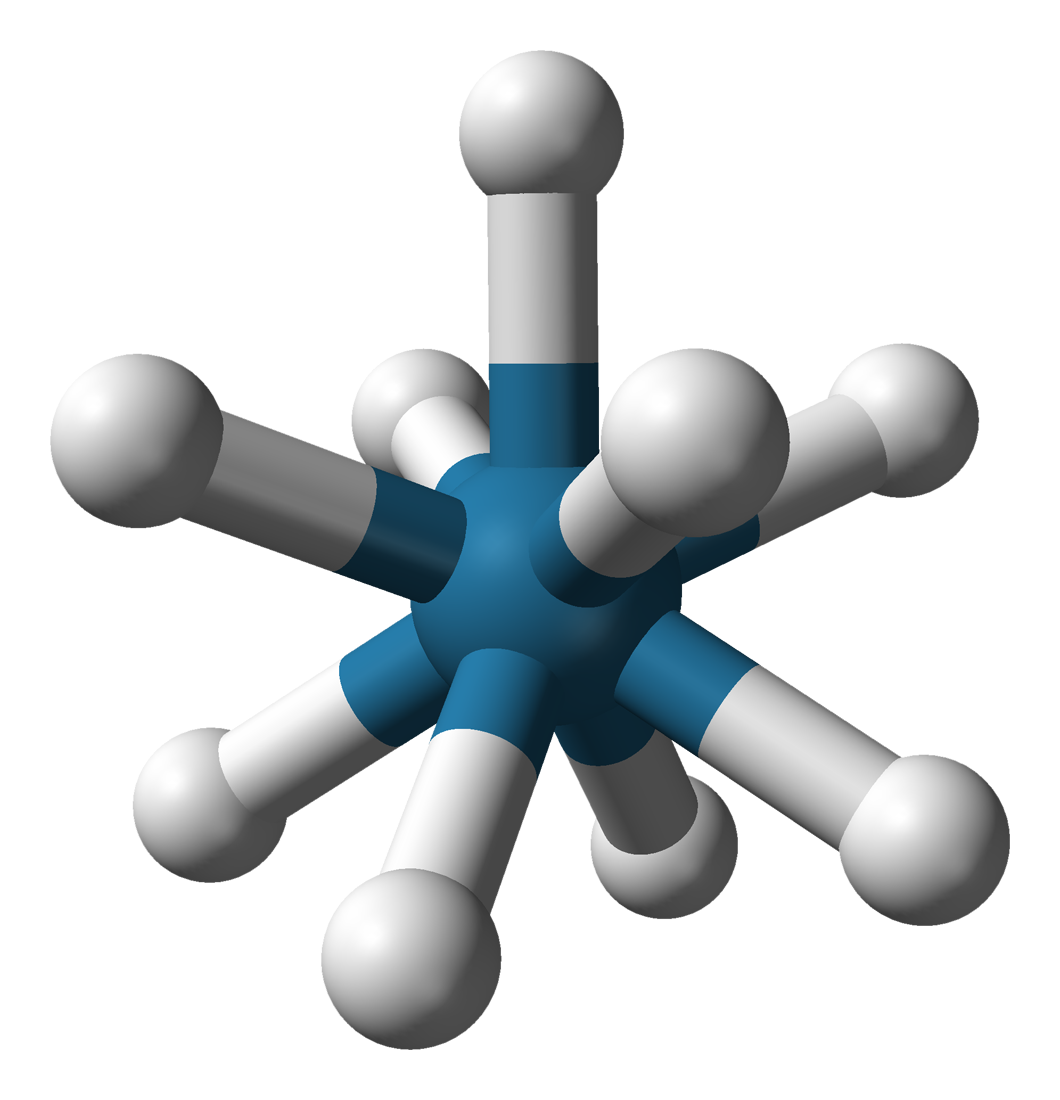
Ti dans la fresnoite Ba2(TiO)Si2O7[14]6 octaèdre 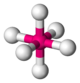
Ti dans l'ion hexaaquatitane(III) Ti(H2O)62+ Na et Cl dans NaCl[15],
Ti dans BaTiO3 (structure pérovskite)[16],
Ni dans la breithauptite NiSb[17]prisme
triangulaire
ou trigonal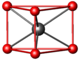
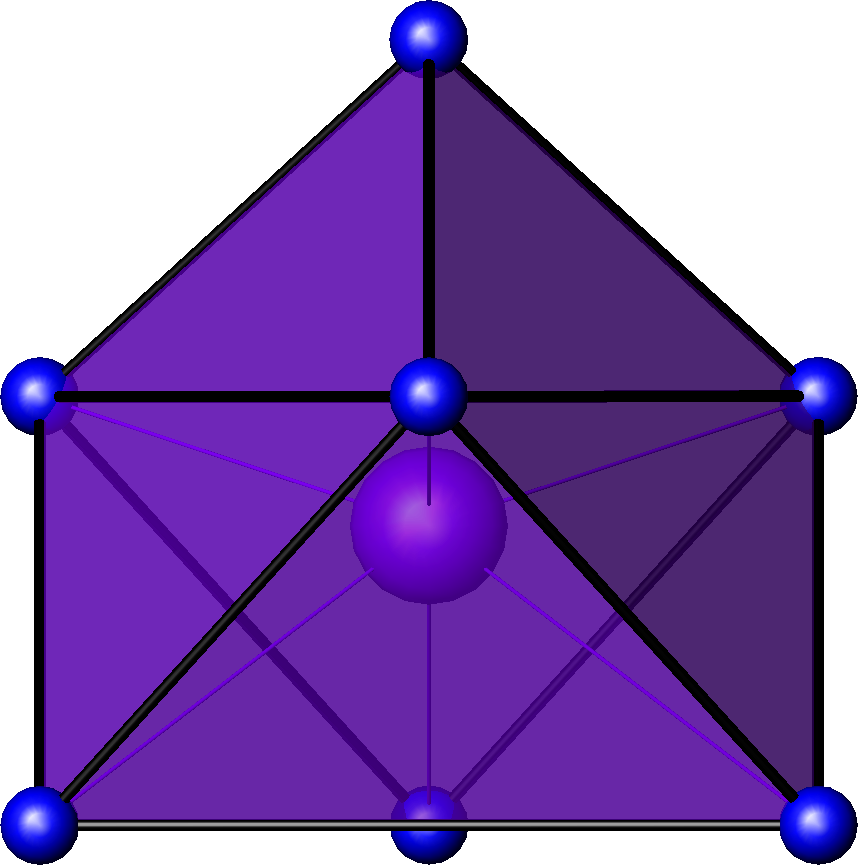
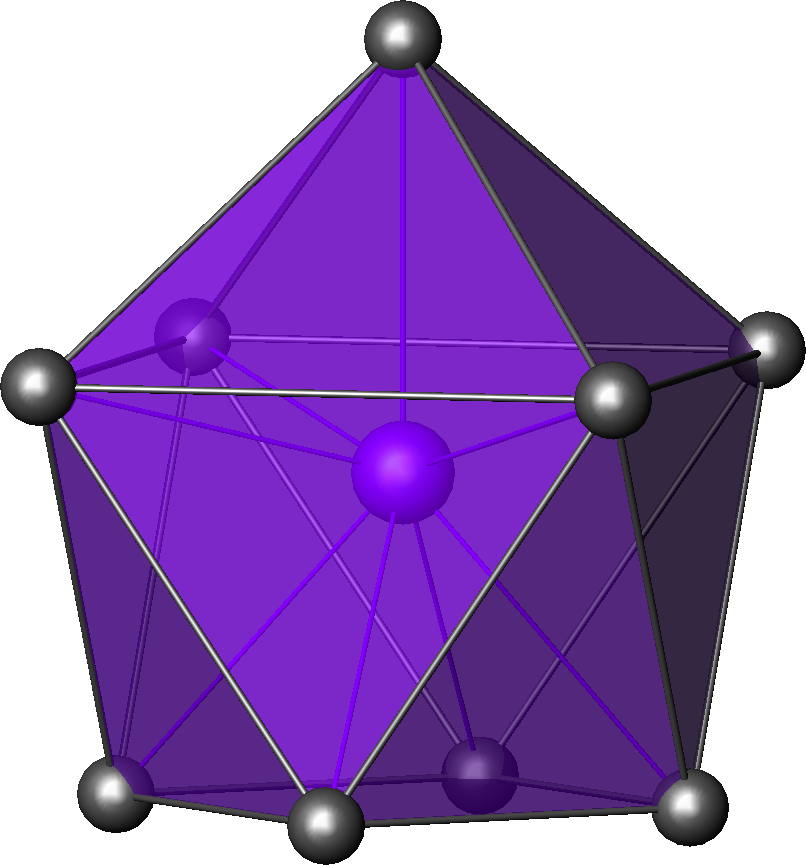
Sb dans la breithauptite 7 bipyramide
pentagonale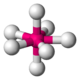
U dans l'ion UO2(H2O)52+[18] Pa dans PaCl5[19] octaèdre à
face coiffée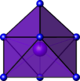
La dans A-La2O3[20] prisme trigonal
à face carrée
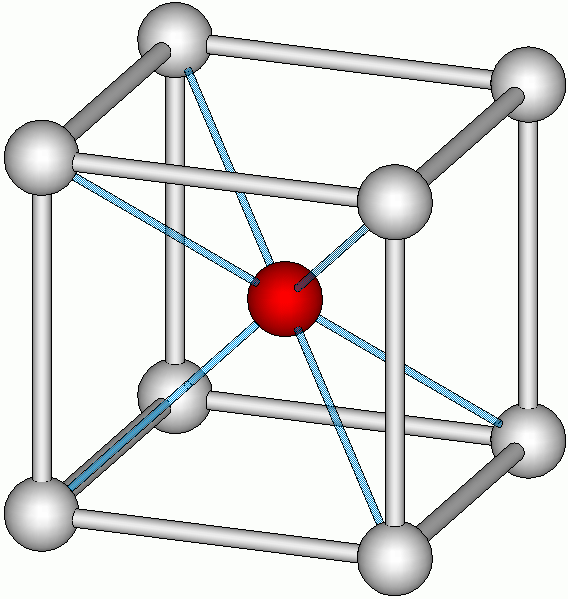
coiffée8 cube 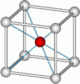
Cs et Cl dans CsCl[21] antiprisme
carré ou
tétragonal
bipyramide
hexagonale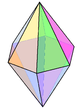
N dans Li3N[22] disphénoïde
adouci
Zr dans K2ZrF6[23] prisme trigonal
à deux faces
triangulaires
coifféesprisme trigonal
à deux faces
carrées coiffées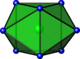
Ca dans CaFe2O4[24] 9 prisme trigonal
à trois faces
carrées coiffées
Pr dans PrZr3F15[25],
Sr dans SrCl2·6H2O[26]antiprisme
carré à face
carrée coiffée
La dans LaTe2[27] 10 antiprisme
carré à faces
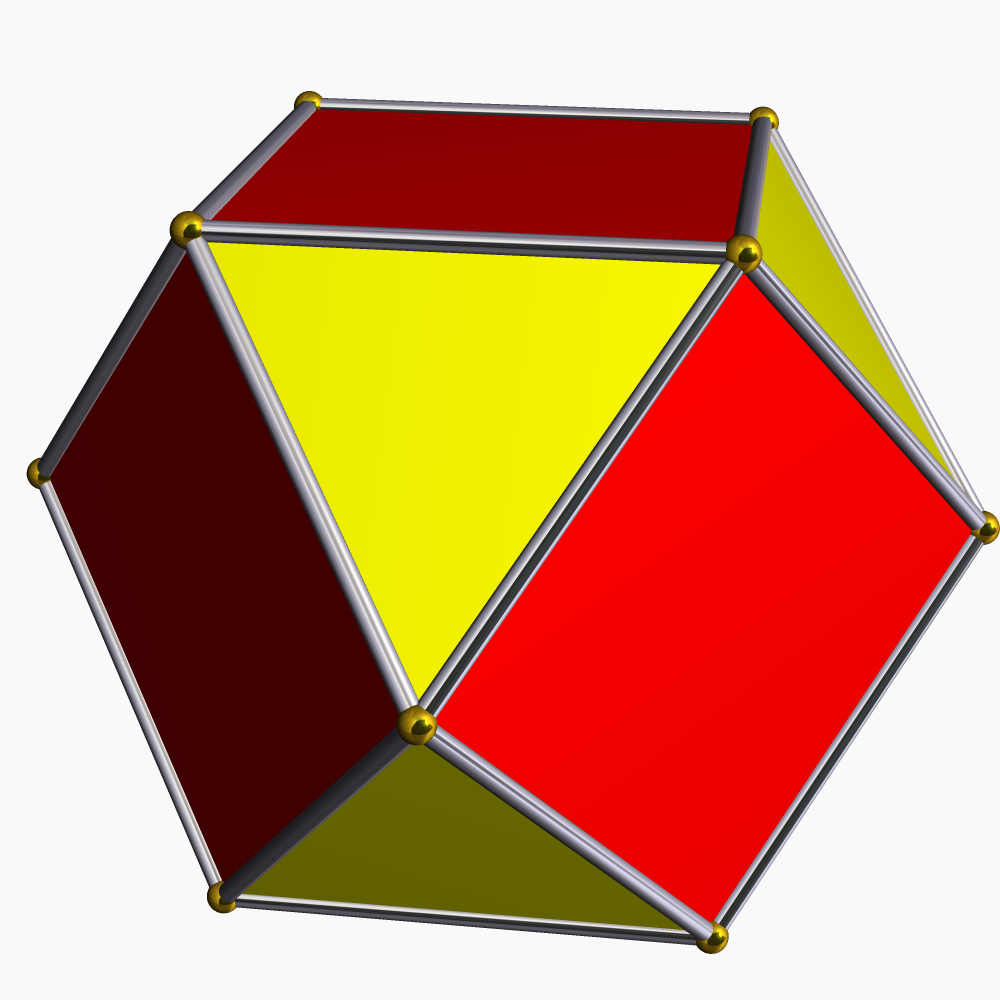
coiffées11 12 icosaèdre 
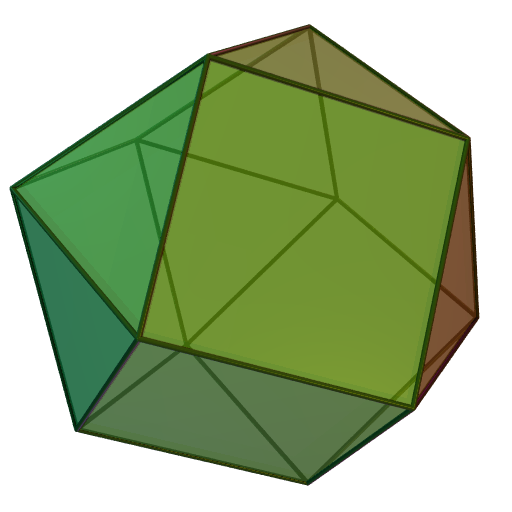
Cr dans Mg3Cr2Al18[28] cuboctaèdre 
Les atomes dans les métaux à structure
de type empilement compact cubique
à faces centréesorthobicoupole
hexagonale ou
anticuboctaèdre
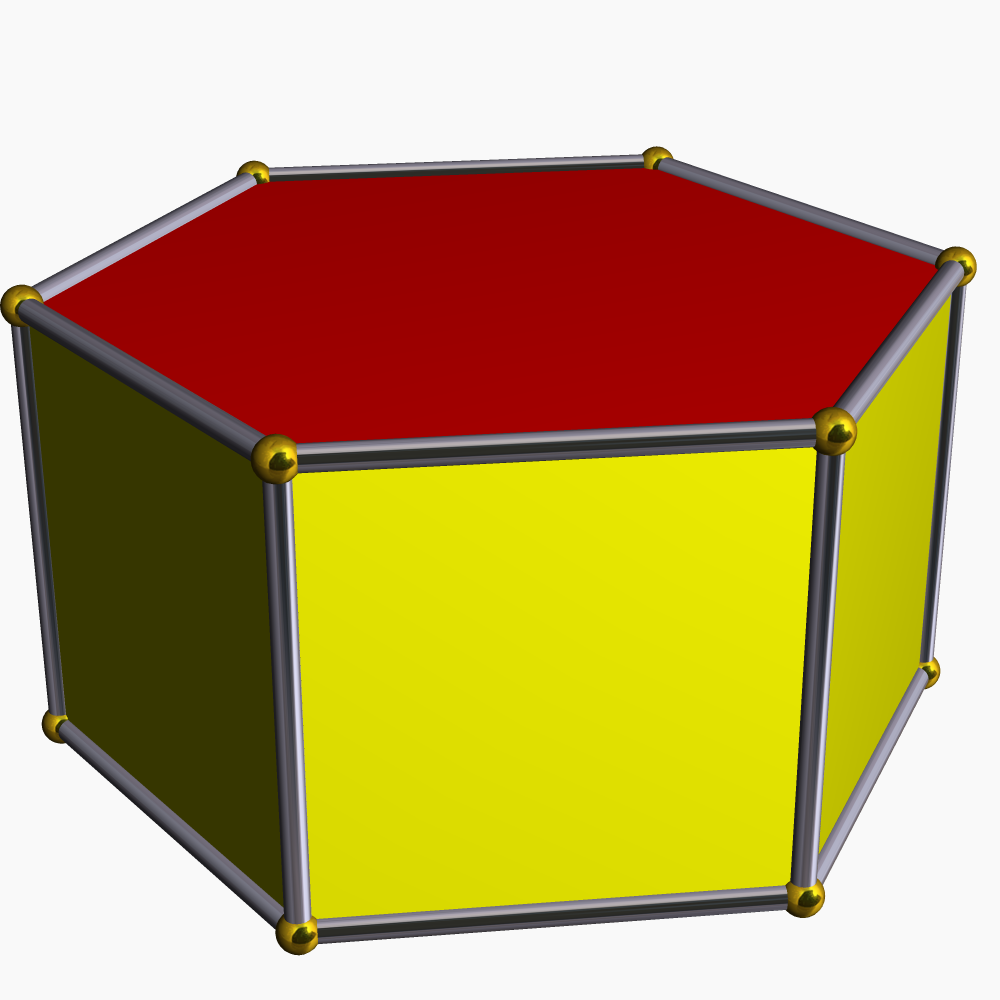
Les atomes dans les métaux à structure
de type empilement compact hexagonalprisme
hexagonal
Rb dans Rb2(VO)2[Si8O19][29] Détermination d'un polyèdre de coordination
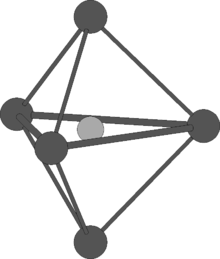 Bipyramide trigonale idéalisée. Les arêtes épaisses correspondent aux angles diédraux de 53,1°, les arêtes plus fines aux angles diédraux de 101,5°. Les faces ne sont pas des triangles équilatéraux.
Bipyramide trigonale idéalisée. Les arêtes épaisses correspondent aux angles diédraux de 53,1°, les arêtes plus fines aux angles diédraux de 101,5°. Les faces ne sont pas des triangles équilatéraux.
Dans la pratique, les polyèdres de coordination dans les matériaux cristallins sont souvent déterminés à partir d'expériences de diffraction de rayons X, de neutrons ou d'électrons. Des méthodes de spectroscopie comme la spectrométrie d'absorption des rayons X peuvent aussi donner des informations sur l'environnement des espèces chimiques, en particulier lorsqu'elles se trouvent en solution liquide ou dans un matériau amorphe.
Dans le cas des complexes métalliques synthétisés en solution, la coordinence n'est complètement établie qu'une fois la structure cristallographique résolue. Il est alors intéressant d'analyser les écarts entre les angles du polyèdre de base (par exemple l'octaèdre) et les angles effectivement observés. Ils sont souvent dus à des contraintes stériques. Il en est de même pour les distances cations - ligands. La déformation des distances pour des ligands de dimension comparable peut être interprétée par l'effet Jahn Teller. C'est le cas fréquent du cuivre(II). La géométrie la plus symétrique en solution, qui est généralement acceptée en bon accord avec les données RMN, n'est qu'une moyenne entre les différentes formes qui s'échangent les unes les autres par vibration de la sphère de complexation.
Les polyèdres de coordination sont en général décrits dans les publications scientifiques par la longueur des liaisons chimiques entre le cation central M et ses ligands X, et par les angles entre ces liaisons. La longueur des liaisons dépend des espèces chimiques considérées : dans un tétraèdre SiO4, les liaisons Si-O ont typiquement une longueur de 1,60 Å[30], alors que les liaisons B-O moyennes dans un tétraèdre BO4 varient entre 1,44 Å et 1,53 Å[31]. Pour cette raison, on utilise d'abord les angles de liaison pour déterminer le polyèdre de coordination. Les longueurs de liaisons sont surtout utiles pour calculer le nombre de coordination effectif d'un atome ou pour rendre compte de la multiplicité de la liaison entre le cation métallique et un ligand.
Lorsqu'il y a déformation du polyèdre de coordination, c'est-à-dire lorsque les angles de liaisons observés s'écartent beaucoup des valeurs attendues pour un polyèdre régulier, il n'est pas toujours évident de reconnaître d'emblée le polyèdre de coordination d'un atome. Par exemple, le polyèdre de coordination CuO5 du cuivre peut être une pyramide tétragonale dans le supraconducteur YBa2Cu3O7[32] ou une bipyramide trigonale dans l'olivénite Cu4(AsO4)2(O)[33], mais il est tellement déformé dans Cu2P2O7 qu'il est à mi-chemin entre les deux[34],[35]. Le calcul des angles diédraux du polyèdre de coordination, entre les normales à deux faces adjacentes, permet dans certains cas difficiles de lever le doute[36]. À chaque arête du polyèdre de coordination correspond un angle diédral. Le tableau suivant donne les angles de liaison et les angles diédraux pour quelques polyèdres de coordination idéalisés, pour lesquels toutes les longueurs de liaison M-X sont égales (les polyèdres de coordination ne sont alors pas forcément réguliers, voir le cas de la bipyramide trigonale).
Polyèdre Angles de liaison X-M-X Angles diédraux carré 90° (×4) 0° (×4) tétraèdre 109,5° (×4) 109,5° (×6) pyramide tétragonale 90° (×8) 0° (×1), 75,7° (×4), 119,8° (×4) bipyramide trigonale 120° (×3), 90° (×6) 53,1° (×3), 101,5 (×6) cube 70,5° (×12) 90° (×12) octaèdre 90° (×12) 70,5° (×12) Facteurs déterminant les polyèdres de coordination
Pour une espèce chimique centrale et ses ligands donnés, le nombre de polyèdres de coordination possibles est restreint par plusieurs facteurs, en rapport avec les règles de Pauling.
Influence des rayons ioniques
Quelques quotients critiques Polyèdre Quotient critique triangle
équilatéral
tétraèdre 
carré,
octaèdre
prisme
trigonal
antiprisme
tétragonal
cube 
icosaèdre 
cuboctaèdre,
anticuboctaèdre
Dans une description géométrique simplifiée, l'espèce chimique centrale (généralement un cation) et ses ligands (généralement des anions) sont considérés comme des boules dures de rayons ioniques rc et ra, respectivement. Cette description est surtout valable pour des ligands monoatomiques. Dans le cas de ligands polyatomiques comme les bipyridines, il pourrait être possible de ne considérer que les atomes en contact direct avec l'espèce chimique centrale ; cependant, la forme de la molécule ligand aura une influence sur la taille du polyèdre de coordination.
Les ligands sont en contact avec l'espèce chimique centrale. Ils peuvent aussi toucher les autres ligands adjacents, mais pas nécessairement. La taille du site formé par les ligands en contact dans un polyèdre de coordination donne une limite inférieure pour la taille du cation qui s'y loge. Un cation ne peut pas se loger dans le site formé par les ligands si sa taille est plus petite que celle du site, car dans ce cas il n'est pas en contact direct avec les ligands : les répulsions électrostatiques entre les ligands l'emportent et la structure cristalline n'est plus stable.
Le calcul du quotient des rayons ioniques q = rc / ra permet de déterminer le polyèdre de coordination probable d'une espèce chimique : il suffit de comparer q aux quotients « critiques » qp pour tous les polyèdres de coordination. Au-delà de qp, le polyèdre de coordination considéré n'est plus stable et il est probable que l'espèce chimique adoptera l'environnement possédant un quotient critique supérieur. Par exemple, dans le cas du quartz SiO2, le cation Si4+ a un rayon ionique rc de 0,41 Å et l'anion O2– un rayon ionique ra de 1,40 Å[37], le quotient q vaut 0,293 et le polyèdre de coordination du silicium est donc un tétraèdre SiO4.
Les polyèdres de coordination à haut quotient critique correspondent généralement à une grande coordinence, cependant, la coordinence n'augmente pas forcément avec le quotient critique : le carré (coordinence 4) et l'octaèdre (coordinence 6) possèdent le même quotient critique.
Le quotient critique qp est une caractéristique du polyèdre de coordination considéré et peut être calculé exactement de façon simple.
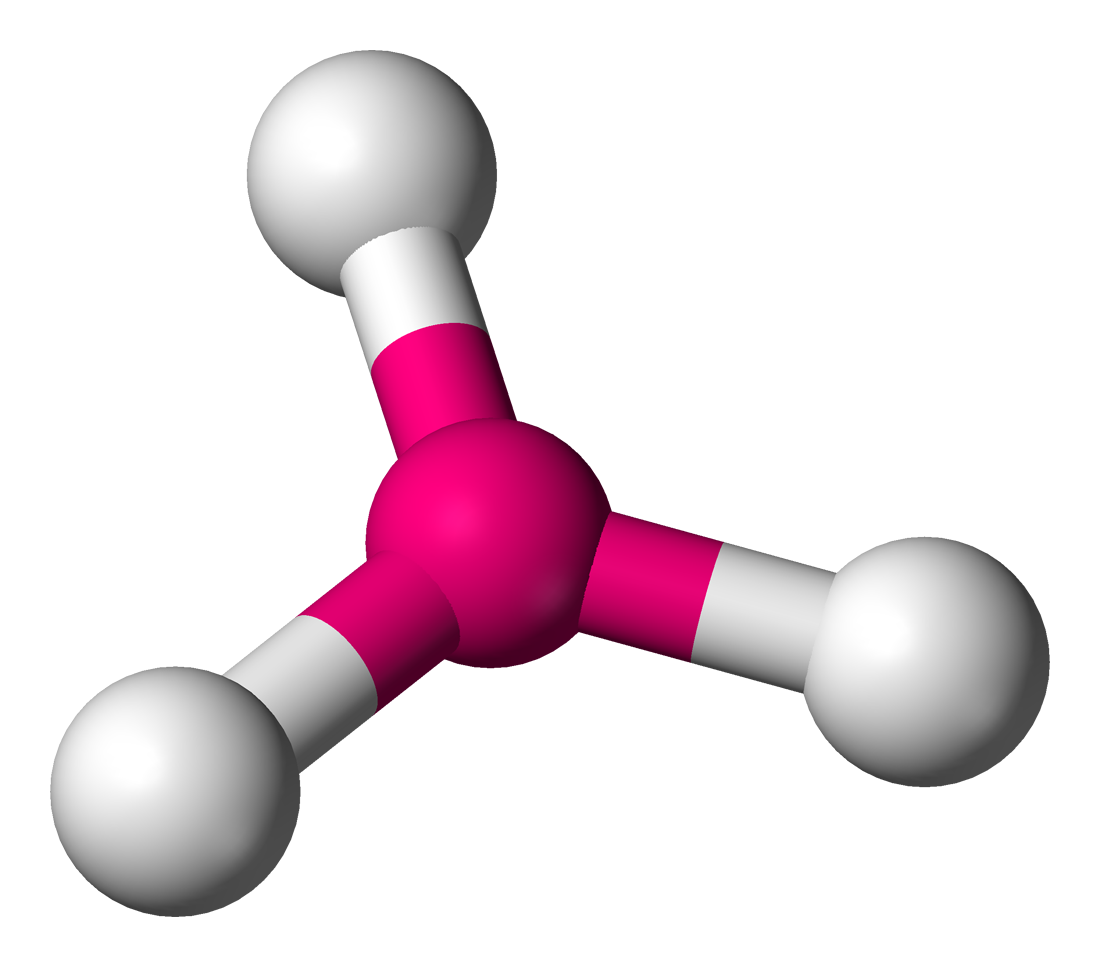
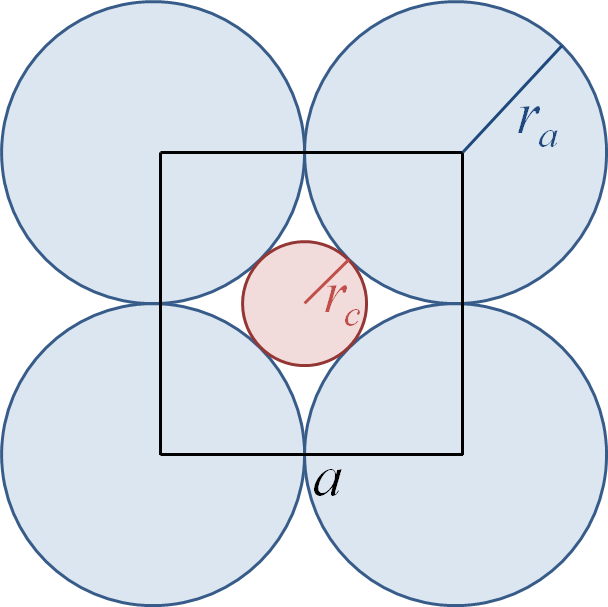
 Coordination de type plan carré.
Coordination de type plan carré.Considérons par exemple le cas de la coordination de type plan carré.
Les centres des ligands constituent un carré de côté a. Il suffit de déterminer l'arête du polyèdre de coordination et sa demi-diagonale en fonction des rayons ioniques ra et rc :
En remplaçant a par son expression en fonction de ra, on obtient
d'où le quotient critique
La méthode de calcul du quotient critique présentée ici pour le carré fonctionne également pour les autres polyèdres de coordination.
Influence des degrés d'oxydation
Le changement du degré d'oxydation d'une espèce chimique a des conséquences sur son polyèdre de coordination. L'effet du degré d'oxydation sur l'environnement chimique n'est pas trivial. D'une part, un cation à haut degré d'oxydation aura tendance à attirer un grand nombre d'anions afin de compenser les charges électrostatiques. D'autre part, le rayon ionique d'un cation diminue lorsqu'il perd plus d'électrons, favorisant un polyèdre de coordination à plus faible quotient critique. Enfin, pour un même degré d'oxydation, une espèce chimique peut avoir plusieurs polyèdres de coordination différents.
Il existe cependant des corrélations entre degré d'oxydation et polyèdre de coordination, notamment pour les métaux de transition :
- cas du vanadium[38] :
- le cation V3+ possède une coordination octaédrique d'anions O2– ;
- le cation V4+, de spin 1/2, peut être en coordination pyramidale tétragonale ou octaédrique d'O2– ;
- le cation V5+ peut être en coordination tétraédrique, pyramidale tétragonale ou octaédrique d'O2– ;
- cas du cuivre :
- le polyèdre de coordination du cation Cu+ est linéaire (groupes CuO2 dans la cuprite Cu2O[39], le semi-conducteur YBa2Cu3O6[40]) ou tétraédrique (groupes CuS4 dans la stannite Cu2FeSnS4[2]) ;
- le polyèdre de coordination du cation Cu2+ (spin 1/2) est très souvent un plan carré (groupes CuO4 dans BaCuSi2O6[1]), une pyramide tétragonale (groupes CuO5 dans le supraconducteur YBa2Cu3O7[32]), un octaèdre (groupes CuCl4(H2O)2 dans la mitscherlichite K2CuCl4·2H2O[41]) ou, plus rarement, une bipyramide trigonale (groupes CuO5 dans l'olivénite Cu4(AsO4)2(O)[33]). Dans le cas de Cu2+, l'effet Jahn-Teller conduit à une forte déformation du polyèdre de coordination au-delà du carré.
Pour les complexes en solution, l'influence du degré d'oxydation s'inteprète souvent avec des arguments orbitalaires. Par exemple, la coordinence du cuivre(II) est un octaèdre déformé (élongation de symétrie D4h due à l'effet Jahn Teller). C'est le cas par exemple de Cu(NH3)4(H2O)22+. En revanche, le cuivre(I) a une coordinence linéaire. C'est le cas des organocuprates Cu(CH3)2−. Il en est de même pour l'argent(I) avec le réactif de Tollens Ag(NH3)2+ et les complexes d'or(I).
Déformation d'un polyèdre de coordination
La déformation d'un polyèdre de coordination MXn est donnée principalement par la déviation des angles de liaison X-M-X par rapport à ceux des polyèdres idéalisés et par la distribution des longueurs de liaison, mais aussi par la distribution des distances entre les ligands.
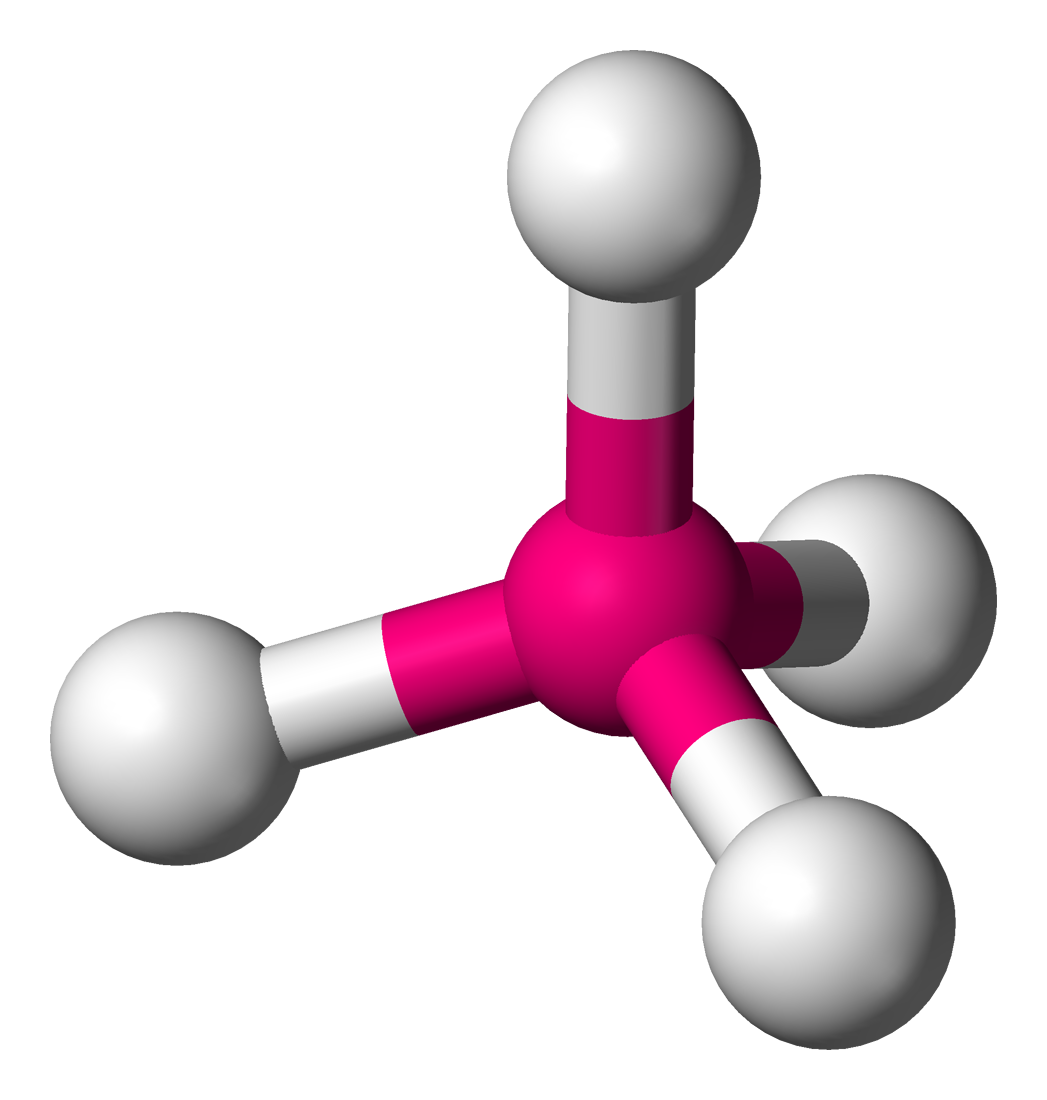
Coordination tétraédrique
Trois paramètres sans dimension sont couramment utilisés pour décrire la déformation d'un tétraèdre MX4[42] :
- BLD (déformation des longueurs de liaison, bond length distortion en anglais) :
-
- où (M–X)i est la longueur de liaison entre l'espèce chimique centrale M et le ligand X, <M–X> est la moyenne des longueurs de liaison du tétraèdre et n est le nombre de liaisons dont la longueur dévie de la longueur moyenne de plus d'une fois et demi l'écart-type sur sa valeur ;
- AD (déformation angulaire, angular distortion) :
-
- avec
- où α0 est l'angle de liaison dans le tétraèdre idéalisé, la somme sur i se faisant sur les trois angles adjacents à la liaison considérée ;
- ELD (déformation des côtés du tétraèdre, edge length distortion) :
-
- où n est cette fois le nombre de côtés du tétraèdre, soit 6.
Notes et références
- (en) L.W. Finger, R.M. Hazen et R.J. Hemley, « BaCuSi2O6: A new cyclosilicate with four-membered tetrahedral rings », dans Am. Mineral., vol. 74, no 7-8, 1989, p. 952-955 [texte intégral]
- (en) S.R. Hall, J.T. Szymanski et J.M. Stewart, « Kesterite, Cu2(Zn,Fe)SnS4, and stannite, Cu2(Fe,Zn)SnS4, structurally similar but distinct minerals », dans Can. Mineral., vol. 16, no 2, 1978, p. 131-137
- Ce tableau est tiré en partie de l'article anglais Coordination geometry dans sa version du 5 novembre 2010.
- (en) J.L. Hoard, « The crystal structure of potassium silver cyanide », dans Zeitschrift für Kristallographie, Kristallgeometrie, Kristallphysik, Kristallchemie, vol. 84, 1933, p. 231-255
- (en) J.K. Nimmo et B.W. Lucas, « A neutron diffraction determination of the crystal structure of α-phase potassium nitrate at 25 degrees C and 100 degrees C », dans J. Phys. C: Solid State Phys., vol. 6, no 2, 1973, p. 201-211 [lien DOI]
- (en) H. Chessin, W.C. Hamilton et B. Post, « Position and thermal parameters of oxygen atoms in calcite », dans Acta Cryst., vol. 18, no 4, 1965, p. 689-693 [lien DOI]
- Miessler et Tarr, p.423.
- (de) R. Brill, C. Hermann et Cl. Peters, « Studien über chemische Bindung mittels Fourieranalyse III. Die Bindung im Quarz », dans Naturwissenschaften, vol. 27, no 40, 1939, p. 676-677 [texte intégral, lien DOI]
- Miessler et Tarr, p.424.
- (en) S. Åsbrink et L.-J. Norrby, « A refinement of the crystal structure of copper(II) oxide with a discussion of some exceptional e.s.d.'s », dans Acta Cryst. B, vol. 26, no 1, 1970, p. 8-15 [lien DOI]
- (en) A.R.H.F Ettema et R.A de Groot, « Bandstructure calculations of the hexagonal and cubic phases of K3Sb », dans J. Phys.: Condens. Matter, vol. 11, no 3, 1999, p. 759-766 [texte intégral, lien DOI]
- Miessler et Tarr, p.375.
- J.-C. Bouloux et J. Galy, « Structure cristalline de l'hypovanadate CaV4O9 », dans Acta Cryst. B, vol. 29, no 6, 1973, p. 1335-1338 [lien DOI]
- (en) Paul B. Moore et S. John Louisnathan, « The crystal structure of fresnoite, Ba2(TiO)Si2O7 », dans Z. Kristall., vol. 130, no 1-6, 1969, p. 438-448 [texte intégral, lien DOI]
- (de) M. Straumanis et A. Ieviņš, « Die Gitter konstanten des NaCl und des Steinsalzes », dans Zeitschrift für Physik, vol. 102, no 5-6, 1936, p. 353-359 [lien DOI]
- (en) F.J. Gotor, C. Real, M.J. Dianez et J.M. Criado, « Relationships between the Texture and Structure of BaTiO3 and Its Tetragonal → Cubic Transition Enthalpy », dans J. Solid State Chem., vol. 123, no 2, 1996, p. 301-305 [lien DOI]
- (en) Robert Leubolt, Herbert Ipser, Peter Terzieff et Kurt L. Komarek, « Nonstoichiometry in B8-Type NiSb », dans Z. Anorg. Allg. Chem., vol. 533, no 2, 1986, p. 205-214 [lien DOI]
- R. Souane (2005) Synthèse et propriétés complexantes vis-à-vis de l’ion uranyle de dérivés carboxyliques du p-tert-butyl-calix[6]arène. Thèse de doctorat de l'université L. Pasteur de Strasbourg. p.90 http://scd-theses.u-strasbg.fr/933/01/these-souane.pdf
- (en) R.P. Dodge, G.S. Smith, Q. Johnson et R.E. Elson, « The crystal structure of protactinium pentachloride », dans Acta Cryst., vol. 22, no 1, 1967, p. 85-89 [lien DOI]
- P. Aldebert et J.P. Traverse, « Étude par diffraction neutronique des structures de haute température de La2O3 et Nd2O3 », dans Materials Research Bulletin, vol. 14, no 3, mars 1979, p. 303-323 [lien DOI]
- (en) Wheeler P. Davey, « Precision Measurements of Crystals of the Alkali Halides », dans Phys. Rev., vol. 21, no 2, 1923, p. 143-161 [lien DOI]
- (en) Takeshi Asai, Kunio Nishida et Shichio Kawai, « Synthesis and ionic conductivity of CuxLi3−xN », dans Materials Research Bulletin, vol. 19, no 10, octobre 1984, p. 1377-1381 [lien DOI]
- (de) R. Hoppe et B. Mehlhorn, « Die Kristallstruktur von K2ZrF6 », dans Z. Anorg. Allg. Chem., vol. 425, no 3, 1976, p. 200-208 [lien DOI]
- (en) B.F. Decker et J.S. Kasper, « The structure of calcium ferrite », dans Acta Cryst., vol. 10, no 4, 1957, p. 332-337 [lien DOI]
- (en) J.P. Laval et A. Abouz, « Crystal chemistry of anion-excess ReO3-related phases: Crystal structure of β-PrZr3F15 », dans J. Solid State Chem., vol. 96, no 2, 1992, p. 324-331 [lien DOI]
- (en) P.A. Agron et W.R. Busing, « Calcium and strontium dichloride hexahydrates by neutron diffraction », dans Acta Cryst. C, vol. 42, no 2, 1986, p. 141-143 [lien DOI]
- (en) W.B. Pearson, « The Cu2Sb and related structures », dans Z. Kristall., vol. 171, no 1-2, 1985, p. 23-29 [lien DOI]
- (en) S. Samson, « The crystal structure of the intermetallic compound Mg3Cr2Al18 », dans Acta Cryst., vol. 11, no 12, 1958, p. 851-857 [lien DOI]
- (en) Sebastian Prinz, Karine M. Sparta et Georg Roth, « The diphyllosilicate Rb2(VO)2[Si8O19] », dans Acta Cryst. C, vol. 64, no 3, 2007, p. i27-i29 [lien DOI]
- (de) Th. Hahn, « Silikate - Struktur, Eigenschaften und Technische Nutzung », dans Fakultät für Bergbau und Hüttenwesen der RWTH-Aachen, Technische Mitteilungen, vol. 56, no 12, 1963, p. 501-509
- (de) E. Zobetz, « Geometrische Größen und einfache Modellrechnungen für BO4-Gruppen », dans Z. Kristall., vol. 191, no 1-2, 1990, p. 45-57 [lien DOI]
- C. Michel et B. Raveau, « Les oxydes A2BaCuO5 (A = Y, Sm, Eu, Gd, Dy, Ho, Er, Yb) », dans J. Solid State Chem., vol. 43, no 1, 1982, p. 73-80 [lien DOI]
- (en) Richard D. Adams, Ralph Layland et Christophe Payen, « The Synthesis, Crystal Structures and Magnetic Properties of Cu4(AsO4)2(O) and Ba2Cu7(AsO4)6 », dans Chem. Ber./Recueil, vol. 130, no 1, 1997, p. 63-67 [lien DOI]
- (en) B.E. Robertson et C. Calvo, « The Crystal Structure and Phase Transformation of α-Cu2P2O7 », dans Acta Cryst., vol. 22, no 5, 1967, p. 665-672 [lien DOI]
- (en) Karine Sparta, Achim Löffert, Christoph Gross, Wolf Aßmus et Georg Roth, « Single crystal X-ray structure analysis of the spin ladder compounds SrCu2O3 and Sr2Cu3O5 between 300 K and 100 K », dans Z. Kristall., vol. 221, no 12, 2006, p. 782-787 [lien DOI]
- (en) E.L. Muetterties et L.J. Guggenberger, « Idealized Polytopal Forms. Description of Real Molecules Referenced to Idealized Polygons or Polyhedra in Geometric Reaction Path Form », dans J. Am. Chem. Soc., vol. 96, no 6, 1974, p. 1748-1756 [lien DOI]
- (en) R.D. Shannon, « Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides », dans Acta Cryst. A, vol. 32, 1976, p. 751-767 [lien DOI]
- (en) M. Schindler, F.C. Hawthorne et W.H. Baur, « Crystal Chemical Aspects of Vanadium: Polyhedral Geometries, Characteristic Bond Valences, and Polymerization of (VOn) Polyhedra », dans Chem. Mater., vol. 12, no 5, 2000, p. 1248-1259 [texte intégral, lien DOI]
- (en) R. Restori et D. Schwarzenbach, « Charge density in cuprite, Cu2O », dans Acta Cryst. B, vol. 42, no 3, 1986, p. 201-208 [lien DOI]
- (en) Mary F. Garbauskas, R.W. Green, R.H. Arendt et J.S. Kasper, « X-ray investigation of barium yttrium cuprate YBa2Cu3O6 », dans Inorg. Chem., vol. 27, no 5, 1988, p. 871-873 [lien DOI]
- (en) B. Renner et G. Lehmann, « Correlation of angular and bond length distortions in TO4 units in crystals », dans Z. Kristall., vol. 175, no 1-2, 1986, p. 43-59 [lien DOI]
Voir aussi
Articles connexes
 Portail de la chimie
Portail de la chimie Portail des sciences des matériaux
Portail des sciences des matériaux Portail des minéraux et roches
Portail des minéraux et roches




![\begin{array}{l} a = 2r_a \\[1ex] \displaystyle{\frac{a\sqrt{2}}{2}} = r_a+r_c \end{array}](5/79583022a8baade5c36b2b0b5f4cfd29.png)






Wikimedia Foundation. 2010.