- Transesterification
-
Ester
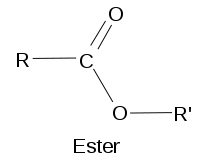 Séquence de la fonction ester.
Séquence de la fonction ester.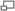
La fonction ester est constituée d'un atome de carbone lié simultanément à un atome d'oxygène par double liaison et à un groupement OR. Les esters sont des dérivés des acides carboxyliques. Les fonctions esters se retrouvent dans de nombreuses molécules biologiques, notamment les triglycérides.
Plusieurs esters ont une odeur agréable et ils sont souvent à l'origine de l'arôme naturel des fruits. Ils sont aussi beaucoup utilisés pour les arômes synthétiques et dans la parfumerie.
Sommaire
Nomenclature
Le nom d'un ester comporte deux termes :
- le premier, qui se termine en -oate, désigne la chaîne principale qui provient de l'acide carboxylique. Elle est liée au carbone et est numérotée quand c'est nécessaire à partir de celui-ci.
- le second, qui se termine en -yle, est le nom du groupe alkyle provenant de l'alcool. Cette chaîne est numérotée à partir de l'atome de carbone lié à l'atome d'oxygène de la fonction ester.
Exemple : le pentanoate d’éthyle
 Structure moléculaire schématique du pentanoate d’éthyle :
Structure moléculaire schématique du pentanoate d’éthyle :
- à gauche, le groupement pentanoate (chaîne de 5 atomes de carbone, le premier au centre de la figure étant doublement lié à un atome d’oxygène en groupement oxyle) ; ce carboxyle provient de l’estérification de l’acide pentanoïque ;
- à droite, le groupement éthyle (chaîne de 2 atomes de carbones, le premier au centre de la figure étant simplement lié à un atome d’oxygène qui assure aussi la liaison faible entre les deux groupements) ; cet alkyle provient de l’estérification de l’éthanol.Synthèse des esters : estérification
Synthèse à partir d'acides carboxyliques
Réaction
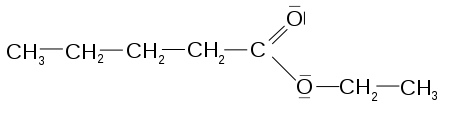
La réaction chimique qui produit un ester et H2O, à partir d'un alcool et d'un acide carboxylique (R-COOH) est appelée estérification ou réaction de Fischer. L'équation générale de cette réaction est :
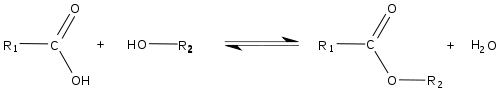
La réaction inverse est une hydrolyse. Les réactions dans les deux sens sont très lentes en absence d'un catalyseur, le proton « libre » (ion hydrogène H +, se présentant sous la forme d'un ion hydronium H3O+ en solution aqueuse) provenant soit d’un acide fort (molécule qui possède un proton se dissociant en solution aqueuse), ou de l’eau dans laquelle l’acide carboxylique est en solution (l'estérification est d'autant plus lente que le pH de cette solution est élevé puisque pH=-log[H+]).
Propriétés
Cette réaction est renversable (saponification) et réversible (la réaction inverse est une "rétro-estérification"), lente et limitée (à cause justement de sa réaction inverse, l'hydrolyse). Elle est aussi quasi athermique[1] (elle ne dégage ni absorbe de la chaleur).
Comme la réaction est athermique, une variation de la température n'a aucune influence sur le rendement (Loi expérimentale de Van't Hoff). De même, une variation de la pression n'entraîne aucun déplacement de l'équilibre (vu que dans quasiment tous les cas, les réactifs et les produits sont des liquides, Loi expérimentale de Le Châtelier). Au mieux, une augmentation de la température accélère la réaction et permet d'atteindre plus rapidement la limite de l'équilibre d'estérification.
Rendement
Le rendement dépend très peu de la nature de l'acide carboxylique utilisé. Il dépend surtout de la classe de l'alcool utilisé : pour des réactifs introduits en quantités équimolaires, il est de 67 % avec un alcool primaire (méthanol par exemple), de 60 % avec un alcool secondaire (ex : isopropanol ou propan-2-ol) et de seulement 5% si l'alcool est tertiaire [2](ex : tertbutanol ou 2-méthylpropan-2-ol).
Note historique: ces résultats, expérimentaux, (athermiticité, rendement dépendant de la classe de l'alcool et peu de l'acide carboxylique, etc.) sont dus en grande partie aux travaux de Marcellin Berthelot et d'Armand Péan de Saint-Gilles (Mémoire de Berthelot et Péan de Saint-Gilles, 1861).
Pour augmenter le rendement, il existe différentes méthodes :
- Augmenter la quantité du réactif en excès (en général le moins cher), ce qui modifiera le taux d'avancement final, donc le rendement.
- Toutes les méthodes permettant d'empêcher l'hydrolyse de se produire, et donc permettant de déplacer l'équilibre dans le sens direct (estérification). On peut au choix :
- Distiller l'ester au fur et à mesure de sa formation, s'il est le plus volatil (ce qui est souvent le cas).
- Éliminer l'eau (pour éviter son ionisation source de protons catalyseurs inverses). Pour cela deux méthodes sont possibles :
- Réaliser un entraînement à la vapeur, en ajoutant au système réactionnel un solvant relativement volatil et formant avec l'eau un hétéroazéotrope. On choisit en général du cyclohexane ou du toluène, et on utilise pour cette méthode l'appareil de Dean Stark.
- Incorporer au mélange réactionnel une substance déshydratante. Cela pose plus de problèmes, car d'une part, même s'il est mis en excès, toute l'eau peut ne pas être consommée. D'autre part, il faut par la suite séparer l'ester de ce produit, ce qui peut entraîner des complications (et en plus faire baisser le rendement).
Cinétique
La réaction non catalysée est assez lente (pour atteindre le rendement maximal, il faut plusieurs mois). La vitesse évolue aussi selon la classe des alcools : elle décroît quand on passe d'un alcool primaire à un alcool secondaire, puis à un alcool tertiaire. Quoi qu'il en soit, on cherche donc des moyens d'accélérer la réaction :
- Augmentation de la température : si elle n'a aucune influence sur le rendement, elle améliore grandement la cinétique.
- Utilisation d'un catalyseur. On utilise pour cela un acide, qui permet d'augmenter le caractère électrophile du groupe carboxyle.
La plupart des réactions permettent d'utiliser de l'acide sulfurique, mais cela n'est pas le cas de toutes (certains composés ne "supportent" pas le "traitement de choc" à l'acide sulfurique à chaud, puissant oxydant, qui risque donc d'oxyder l'alcool, ou même de le déshydrater). On peut utiliser aussi HCl anhydre, ou un acide moins fort, comme l'acide phosphorique, H3PO4, ou l'acide paratoluènesulfonique (APTS), voire, s'il est assez fort (exemple : l'acide méthanoïque, pKA=3,77), l'acide carboxylique réactif ( ⇒ autocatalyse), mis en excès. Cela présente un double avantage : il catalyse ainsi la réaction, et en plus, comme on l'a vu précédemment, cela permet d'améliorer le rendement.
Mécanisme
En fonction de la classe de l'alcool, il existe différents mécanismes.
Le premier mécanisme présenté est valable pour les alcools primaires et secondaires, le second pour les alcools tertiaires.
On prend ici le cas général et on choisit pour catalyseur H+.
Alcools primaires et secondaires
Ce mécanisme se décrit en cinq étapes (dont deux équilibres de protonation-déprotonation rapides).
Première étape : protonation de l'acide carboxylique. Deux possibilités se présentent :
- la protonation du groupe carbonyle
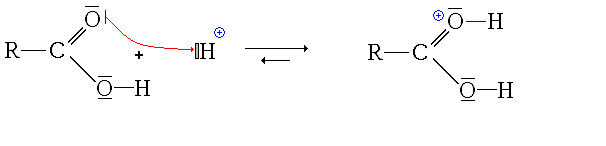
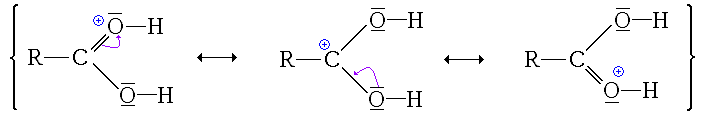
Ici, l'ion formé est stabilisé par mésomérie :
- la protonation du groupe hydroxyle
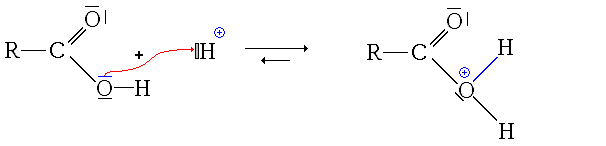
Ici non seulement l'ion formé (ion acyloxonium) ne possède pas de forme mésomère qui le stabilise, mais en plus cet état ne permet pas de poursuivre la réaction. Comme en plus cette réaction est un équilibre, les éventuelles formes protonées au niveau du groupement hydroxyle sont consommées pour former l'autre forme protonée qui sera elle consommée par les étapes suivantes (déplacement de l'équilibre, principe de Le Châtelier).
- La première étape est donc la protonation du groupe carbonyle (équilibre rapidement atteint):
- La deuxième étape est l'attaque nucléophile de l'alcool sur le site électrophile de l'acide carboxylique protoné :
remarque: pour cette étape, on est directement parti de la 2e forme mésomère de l'acide protoné, afin de simplifier le mécanisme.
- La troisième étape est le transfert du proton (H+) du groupe issu de l'alcool sur un des groupes hydroxyles (réaction acide-base interne ou prototropie)
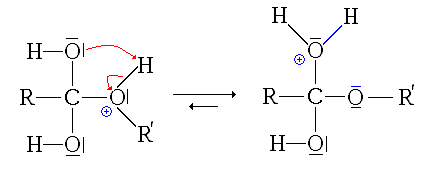
- La quatrième étape, cinétiquement limitante, est le départ d'une molécule d'eau (H2O).
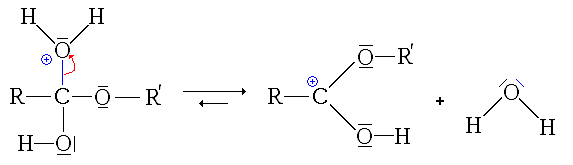
- La dernière étape est une simple déprotonation (restitution du catalyseur)
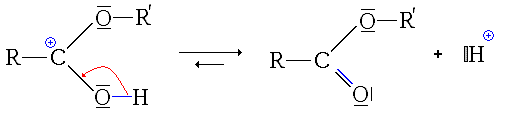
Remarque : le mécanisme a été vérifié en utilisant de l'eau avec un isotope 18O, en suivant la réaction par spectrométrie de masse.
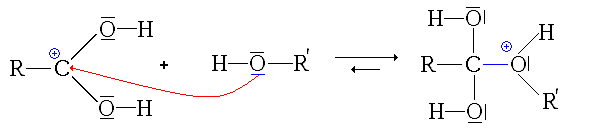
Alcools tertiaires
Ici aussi le mécanisme a lieu en 4 étapes
- Première étape: protonation de l'alcool
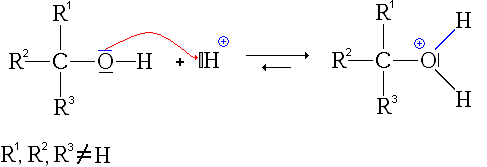
- Deuxième étape: départ de H2O, formation du carbocation tertiaire.
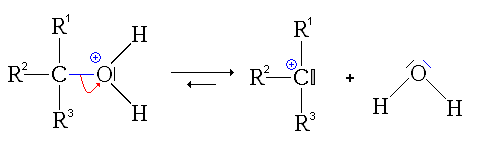
Ces deux étapes (surtout la 2e) sont impossibles avec un alcool primaire ou secondaire, le carbocation formé n'étant pas assez stable.
- Troisième étape : addition du carbocation sur la fonction carbonyle de l'acide carboxylique.
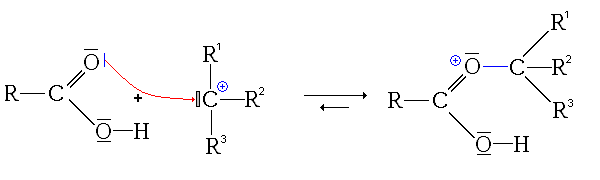
L'intermédiaire ainsi substitué est relativement stable, car il possède plusieurs formes mésomères :
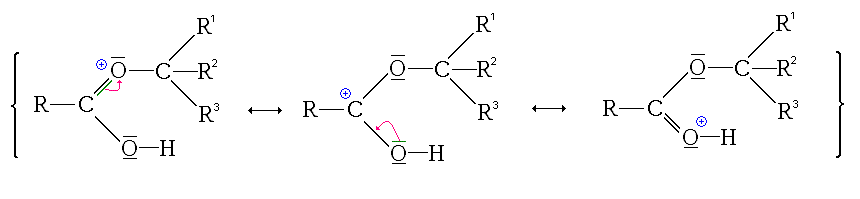
(on part d'ailleurs de la dernière forme mésomère pour la dernière étape)
- Dernière étape: il s'agit juste de la déprotonation de l'intermédiaire précédent (restitution du catalyseur).
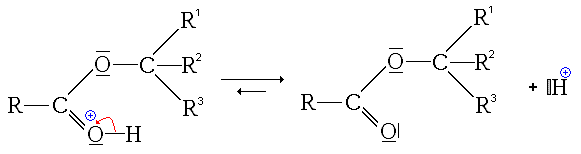
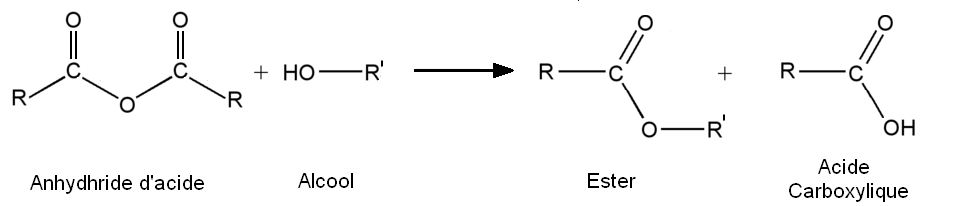
Synthèse à partir de dérivés d'acides carboxyliques
Comme on vient de le voir, la synthèse des esters à partir d'acides carboxyliques présente de nombreux inconvénients : un rendement maximal de l'ordre de 2/3 dans les cas les plus favorables (alcools primaires) et plus que médiocre dans les cas les plus défavorables (5 % pour les alcools tertiaires), une cinétique lente, même catalysée (si la réaction est arrêtée trop rapidement, le rendement baisse encore plus).
Une solution consiste donc à utiliser plutôt des dérivés d'acides, comme les chlorures d'acyles ou les anhydrides d'acides.
Réactions
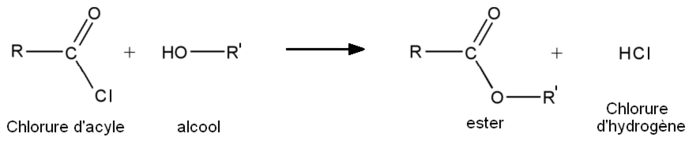
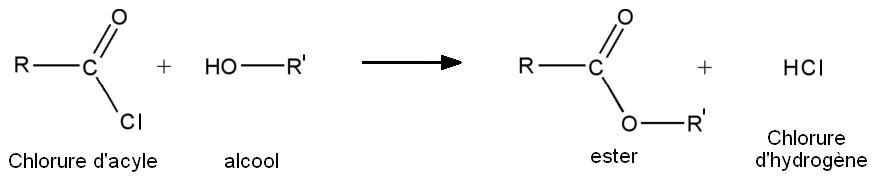
- A partir des chlorures d'acyles :
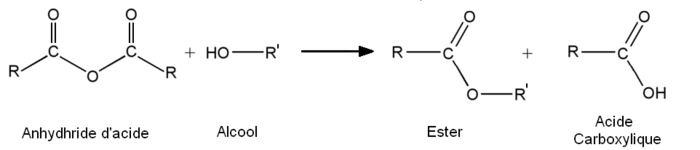
- A partir des anhydrides d'acide :
Propriétés
- Avantages :
- Ces réactions sont totales.
- Elles sont en général relativement rapides, mais nécessitent assez régulièrement une catalyse. Les catalyseurs ne servent qu'à activer et rendre encore plus réactif le dérivé d'acide. Typiquement, dans le cas de l'utilisation d'un chlorure d'acyle, les catalyseurs utilisés sont la pyridine ou la triéthylamine (qui jouent alors le rôle de piège à HCl).
- Inconvénients :
- Les chlorures d'acyle et les anhydrides d'acides réagissent facilement avec l'eau (ils s'hydrolisent très facilement). Cela oblige à prendre des précautions de manipulation (éviter tout contact avec les muqueuses).
- Bien qu'à la base, les réactions soient totales, le rendement n'est pas de 100 %. En effet, une fois la réaction faite, il faut extraire l'ester du milieu, et les étapes pour y arriver sont généralement des équilibres physico-chimiques.
- Les réactions à partir de chlorures d'acyles sont vives : on a besoin de refroidir le mélange.
On a de plus besoin de piéger le HCl formé :
- en utilisant par exemple une base telle la pyridine ou la triéthylamine.
- un piège constitué d'un flacon contenant de la soude.
- Les réactions à partir d'anhydrides sont moins vives et moins rapides (les anhydrides sont moins réactifs).
En industrie, on utilise donc plutôt la voie des acides carboxyliques, plus facile dans la mise en œuvre. Cependant, dans le cas de l'industrie pharmaceutique ou cosmétique, les chlorures d'acyle ou les anhydrides peuvent être utilisés car les produits sont à haute valeur ajoutée.
Mécanismes
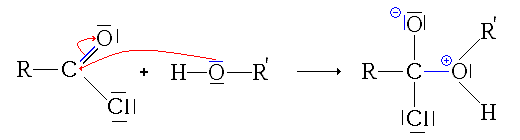
- À partir des chlorures d'acyle
-
- 1re étape : addition nucléophile de l'alcool sur le chlorure d'acyle.
-
- 2e étape : départ de HCl
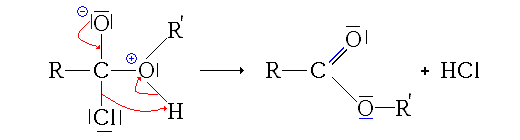
- À partir des anhydrides d'acide
-
- 1re étape : addition nucléophile de l'alcool sur l'anhydride.
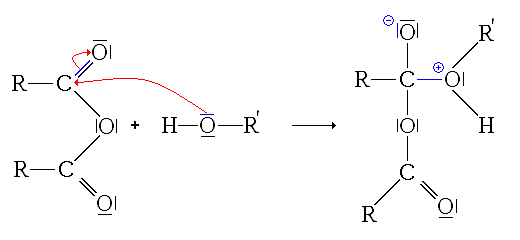
-
- 2e étape : départ de RCOOH
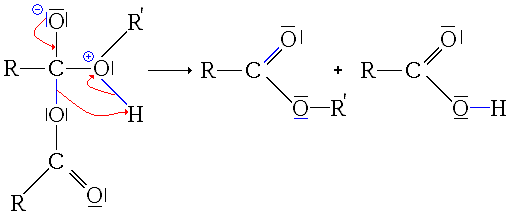
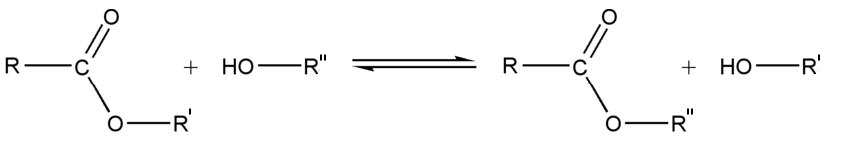
Synthèse à partir d'autres esters (Transestérification)
La transestérification transforme un ester et un alcool en un autre ester et autre alcool. Un acide ou une base est souvent utilisé comme catalyseur.
Réaction
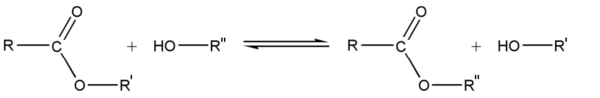
Utilité
La transestérification est utilisée dans la fabrication du polyester et du biodiesel. C'est aussi le mécanisme qui permet l'épissage des introns lors de la maturation des ARNm.
Utilité des esters
- Outre le fait d'obtenir un ester, utile dans l'industrie agroalimentaire, en parfumerie ou d'autres secteurs industriels, l'estérification est utile, de par son caractère réversible (pour les acides carboxyliques et les alcools, tout du moins), dans le cadre de la protection de fonctions.
Puisque la transformation est réversible, elle permet de protéger soit la fonction alcool, soit la fonction acide carboxylique, soit les deux. En effet, si l'on imagine par exemple que l'on veut protéger un alcool, on le fait réagir avec un acide carboxylique pour former un ester ; on fait la réaction que l'on voulait effectuer ; une fois celle-ci finie, on renverse la réaction d'estérification pour retrouver l'alcool. Il existe deux méthodes pour renverser l'estérification :
- On utilise la même réaction (rétro-estérification) en jouant sur les quantités de matière pour que l'équilibre soit favorable à l'acide carboxylique + l'alcool.
- On utilise la réaction de saponification ou hydrolyse en milieu basique des esters.
Les esters sont aussi un constituant de base dans l'industrie des plastiques. Ils sont à la base d'un des plastiques les plus utilisés, le polyester.
- Il s'agit d'un moyen de former les lactones : estérification intramoléculaire à partir d'un hydroxyacide carboxylique.
- Les esters peuvent être réduits :
- en alcools primaires par l'action du tétrahydruroaluminate de lithium (LiAlH4), dans l'éther diéthylique (éther) ou le THF (tétrahydrofurane)
- en aldéhydes par l'action du DIBAL, dans un solvant non polaire (toluène par exemple), à basse température (-60 °C).
- Quelques esters :
Tableau de certains esters et leurs odeurs
(Ce tableau ne demande qu'à être complété)
Ester Odeur Méthanoate
ou Formiatede méthyle Éthérée d'éthyle Odeur de rhum de butyle Fruitée Éthanoate
ou Acétatede méthyle Fruitée d'éthyle Dissolvant ou fruitée de propyle Poire de butyle Banane ou pomme de pentyle Poire d'hexyle Poire d'heptyle Florale d'octyle Orange de linalyle Lavande, bergamote ou banane de 2-phényléthyle Rose de benzyle Jasmin de vinyle Fruitée, agréable à faible concentration
devient vite âcre et irritante à plus forte concentrationd'isoamyle Banane d'isobutyle
ou β-méthylpropylFruitée et florale de 3-méthylbutyle Banane Propanoate
ou Propionated'éthyle Ananas de butyle Pomme de propyle Fruitée d'isoamyle Abricot, ananas d'isobutyle Éthérée d'isopropyle Fruitée Butanoate
ou Butyratede méthyle Pomme d'éthyle Ananas d'isoamyle Pomme Isopentanoate
ou Isovalératede méthyle Fruitée Salicylate de méthyle Thé des bois, Winter-green de phényle Aromatique d'hexyle Azalée Benzoate de méthyle Orientale (très raffinée : "essence de Niobé") d'éthyle Cerise de benzyle Aromatique Notes
Articles connexes
 Portail de la chimie
Portail de la chimie
Catégories : Groupe fonctionnel | Ester | Olfaction



Wikimedia Foundation. 2010.