- Théorie de l'état de transition
-
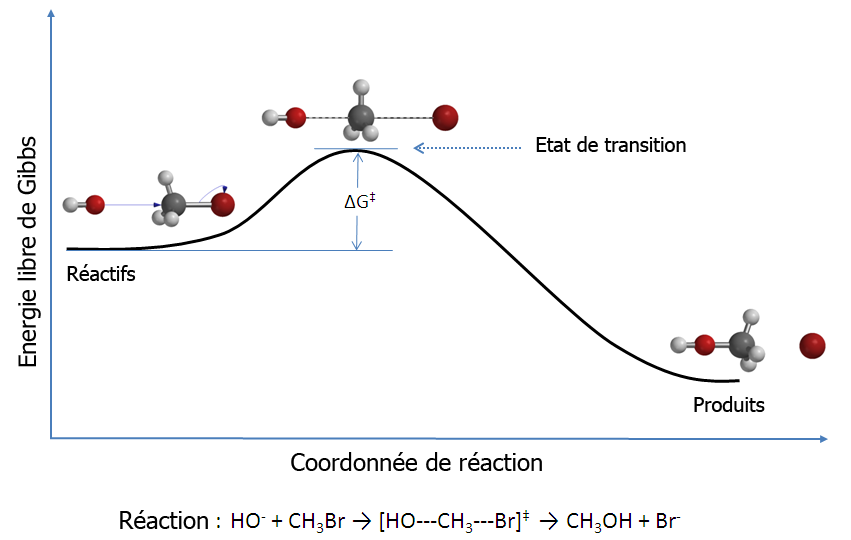
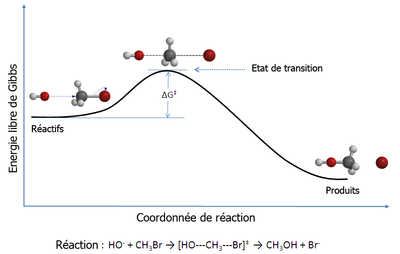 Figure 1 : diagramme de réaction pour une réaction de substitution nucléophile bimoléculaire (SN2) entre le bromométhane et l'anion hydroxyde
Figure 1 : diagramme de réaction pour une réaction de substitution nucléophile bimoléculaire (SN2) entre le bromométhane et l'anion hydroxyde
La théorie de l'état de transition (en anglais transition state theory - TST) a pour objectif d'expliquer les cinétiques de réaction pour des réactions chimiques élémentaires. Cette théorie postule l'existence d'un genre spécial d'équilibre chimique, le quasi-équilibre, entre les réactifs et un complexe de transition activé[1].
La TST est utilisée en premier lieu pour comprendre de manière qualitative le déroulement des réactions chimiques. La théorie a connu moins de succès dans son but initial de calculer des constantes cinétiques absolues pour les réactions, ces calculs nécessitant la connaissance précise des surfaces d'énergies potentielles[2], mais permet de calculer efficacement l'enthalpie standard d'activation (Δ‡H°), l'entropie standard d'activation (Δ‡S°), et l'énergie standard d'activation de Gibbs (Δ‡G°) pour une réaction donnée si sa constante cinétique a été déterminée expérimentalement (la notation ‡ indique que la constante cherchée est à l'état de transition).
Cette théorie a été développée simultanément en 1935 par Henry Eyring, à Princeton, et par Meredith Gwynne Evans et Michael Polanyi à l'Université de Manchester[3],[4].
La théorie de l'état de transition est aussi connue sous les noms de théorie du complexe activé, théorie de la cinétique absolue ou théorie des cinétiques de réaction absolues[5].
Avant le développement de la TST, la loi cinétique d'Arrhenius était largement utilisée afin de déterminer les énergies des barrières de réactions. La loi d'Arrhenius est déduite d'observations empiriques et ne prend pas en compte de considérations mécanistiques, comme par exemple de savoir si un ou plusieurs réactifs intermédiaires sont impliqués dans le passage d'un réactif à un produit[6]. Cependant, un développement plus poussé était nécessaire afin de comprendre les deux paramètres associés à cette loi, le facteur pré-exponentiel (A) et l'énergie d'activation (Ea). La TST, qui conduit à l'équation d'Eyring, permet de déterminer ces deux paramètres ; cependant, 46 ans se sont écoulés entre la publication de la loi cinétique d'Arrhenius en 1889 et celle de l'équation d'Eyring dérivée de la TST en 1935. Durant cette période, de nombreux scientifiques et chercheurs ont contribué de manière significative au développement de la théorie.
Sommaire
Théorie
Les idées de base sur lesquelles s'appuie la théorie de l'état de transition sont les suivantes :
- les cinétiques des réactions sont étudiées par le biais des complexes activés qui se situent aux cols (points-selles) d'une surface d'énergie potentielle. Les détails de la formation de tels complexes importent peu.
- les complexes activés sont en équilibre spécial (quasi-équilibre) avec les molécules réactives.
- les complexes activés peuvent évoluer en produits, ce qui permet à la théorie cinétique de calculer la cinétique de cette conversion.
Développement
Durant le développement de la théorie, trois approches ont été employées, résumées ci-après.
Approche thermodynamique
En 1884, Jacobus van't Hoff proposa la relation indiquant la dépendance en température de la constante d'équilibre d'une réaction réversible :
- A = B
où ΔU est la modification d'énergie interne, K la constante d'équilibre de la réaction, R la constante universelle des gaz parfaits et T la température thermodynamique. En se basant sur l'expérience, Svante Arrhenius proposa en 1889 une expression comparable pour la constante cinétique de réaction, donnée de la manière suivante :
L'intégration de cette équation donne la loi d'Arrhenius :
A était appelé facteur de fréquence (maintenant coefficient pré-exponentiel)et E est considéré comme l'énergie d'activation. Dès le début du XXe siècle, la loi d'Arrhenius était communément admise mais les interprétations physiques de A et de E restaient vagues. Cela conduisit de nombreux chercheurs à s'investir dans la cinétique chimique afin de présenter différentes théories sur le déroulement des réactions afin de lier A et E aux dynamique moléculaires qui en sont responsables[réf. nécessaire].
En 1910, René Marcelin introduisit le concept d'énergie standard d'activation de Gibbs. Son équation peut être écrite comme :
Dans le même temps, les chimistes néerlandais Philip Abraham Kohnstamm, Frans Eppo Cornelis Scheffer et Wiedold Frans Brandsma introduisaient pour la première fois l'enthalpie et l'entropie standard d'activation. Ils proposèrent pour la constante cinétique l'équation :
Cependant, la nature de la constante restait peu claire.
Approche cinétique théorique
Au début de l'année 1900, Max Trautz et William Lewis étudièrent la cinétique de réaction en s'appuyant sur la théorie des collisions, basée sur la théorie cinétique des gaz. La théorie des collisions considère les molécules réactives comme des sphères dures se cognant les unes aux autres mais néglige les modifications de l'entropie.
W. Lewis appliqua cette méthode à la réaction 2HI → H2 + I2 et obtint un bon accord avec les données expérimentales.
Cependant, la même démarche appliquée à d'autres réactions donna lieu à d'importantes différences entre les résultats expérimentaux et théoriques.
Approche en mécanique statistique
La mécanique statistique a joué un rôle significatif dans le développement de la TST. Cependant, l'application de la mécanique statistique à la TST fut développée très lentement étant donné qu'au milieu du XIXe siècle, James Clerk Maxwell, Ludwig Boltzmann et Leopold Pfaundler publièrent de nombreux articles traitant des équilibres et cinétiques réactionnels en termes de dynamique moléculaire et de distribution statistique des vitesses moléculaires.
Il fallut attendre 1912 pour que le chimiste français A. Berthoud utilise la statistique de Maxwell-Boltzmann pour obtenir la constante cinétique.
où a et b sont des constantes liées aux termes d'énergie.
Deux ans après, R. Marcelin produisit une contribution essentielle au problème en considérant l'avancement de la réaction chimique comme le déplacement d'un point dans l'espace des phases. Il appliqua ensuite les méthodes de Gibbs en mécanique statistique et obtint une expression similaire à celle qu'il avait obtenue précédemment à partir de considérations thermodynamiques.
En 1915, une autre contribution importante fut produite par le physicien britannique James Rice. Il conclut sur la base d'une analyse statistique que la constante de réaction est proportionnelle à l'« incrément critique ». Ses idées furent développées plus avant par R. Tolman. En 1919, le physicien autrichien Karl Ferdinand Herzfeld appliqua la mécanique statistique à la constante d'équilibre et la théorie cinétique à la constante cinétique d'une réaction inverse, k-1, pour la dissociation réversible d'une molécule diatomique.
Il obtint l'équation suivante pour la constante cinétique pour cette réaction :
dans laquelle
 est l'énergie de dissociation au zéro absolu, kB la constante de Boltzmann, h est la constante de Planck, T la température thermodynamique et υ la fréquence de vibration de la liaison. Cette expression est très importante car elle fait apparaitre pour la première fois le facteur kBT/h, très important dans la TST, dans une équation cinétique.
est l'énergie de dissociation au zéro absolu, kB la constante de Boltzmann, h est la constante de Planck, T la température thermodynamique et υ la fréquence de vibration de la liaison. Cette expression est très importante car elle fait apparaitre pour la première fois le facteur kBT/h, très important dans la TST, dans une équation cinétique.En 1920, le chimiste américain Richard Chase Tolman développa l'idée de Rice sur l'« incrément critique ». Il conclut que cet incrément critique (connu maintenant sur l'appellation d'énergie d'activation) d'une réaction est égal à l'énergie moyenne de l'ensemble des molécules subissant la réaction moins l'énergie moyenne des molécules réactives.
Surfaces d'énergie potentielle
Le concept de surface d'énergie potentielle fut très important dans le développement de la TST. La base de ce concept fut posée par R. Marcelin. Il postula que l'avancement d'une réaction chimique pouvait être décrit comme un point de la surface d'énergie potentielle avec des coordonnées fonctions des vitesses des atomes et des distances.
En 1931, Henry Eyring et Michael Polanyi décrivirent la surface d'énergie potentielle de la réaction H + H2 → H2 + H. Cette surface est un diagramme tridimensionnel basé sur les principes de mécanique quantique ainsi que sur des données expérimentales sur les fréquences de vibration et les énergies de dissociation.
Un an après cette construction, H. Pelzer et E. Wigner produisirent une contribution importante en suivant l'avancement de la réaction sur la surface d'énergie potentielle. Ce travail discutait pour la première fois du concept de col (ou de point selle) sur la surface d'énergie potentielle. Ils en conclurent que la cinétique de réaction est déterminée par le mouvement du système par ce col.
Origine de l'équation d'Eyring
Article principal : équation d'Eyring.Le point crucial ajouté par Eyring, Polanyi et Evans est que les complexes activés sont en quasi-équilibre avec les réactifs. La constante cinétique est directement proportionnelle à la concentration de ces complexes multipliée par la fréquence (kBT/h) de leur conversion en produits de réaction.
- Approximation de l'état quasi-stationnaire[7]
Il est important de souligner que ce quasi-équilibre est différent de l'équilibre chimique classique, mais qu'il peut être décrit en utilisant le même traitement thermodynamique. Soit la réaction :
 Figure 2 : diagramme d'énergie potentielle.
Figure 2 : diagramme d'énergie potentielle.où l'équilibre est atteint entre toutes les espèces du système, y compris les complexes activés, [AB]‡. En utilisant la mécanique statistique, la concentration de [AB]‡ peut être calculée en fonction des concentrations de A et B.
La TST postule que même lorsque les réactifs et les produits ne sont pas en équilibre entre eux, les complexes activés sont en quasi-équilibre avec les réactifs. Comme indiqué figure 2, à tout instant, il existe quelques complexes activés, certains étant des réactifs dans l'instant précédent, étant désignés par [ABl]‡ (lorsqu'ils sont déplacés de gauche à droite). Le reste d'entre eux étaient des produits dans l'instant précédant, [ABr]‡. Lorsque le système est en équilibre complet, les concentrations en [ABl] ‡ et [ABr]‡ sont égales, donc chaque concentration est égale à la moitié de la concentration totale en complexes activés :
![[AB_\mathrm{r}]^{\ddagger} = \frac{1}{2}[AB]^{\ddagger}](0/040142e809895455493c45b82ac49683.png) et
et ![[AB_\mathrm{l}]^{\ddagger} = \frac{1}{2}[AB]^{\ddagger}](2/002dc21510369ee758792056c5faef92.png)
Si les molécules produit sont subitement retirées du système réactionnel, le flux des complexes activés provenant des produits ([ABr]‡ ) sera interrompu ; cependant, il existera toujours un flux provenant des réactifs. Ainsi, le postulat est que la cinétique du flux de la gauche vers la droite n'est pas influencé par le retrait des produits. En d'autres termes, les flux dans les deux directions sont indépendants l'un de l'autre.
Dans le cadre de la TST, il est important de comprendre que lorsque des complexes activés sont dits en équilibre avec les réactifs, on considère seulement les complexes activés ([ABl] ‡) qui étaient des réactifs dans l'instant immédiat.
La constante d'équilibre K‡o pour le quasi-équilibre peut être écrite :
La concentration de l'état de transition AB‡ est donc :
L'équation cinétique de production du produit est alors :
Où la constante cinétique est donnée par :
k‡ est directement proportionnelle à la fréquence du mode de vibration responsable de la conversion du complexe activé en produit ; ce mode a pour fréquence ν. Chaque vibration ne conduit pas forcément à la formation d'un produit. Une constant proportionnelle κ, appelée coefficient de transmission, est donc introduite pour prendre en compte cet effet. k‡ peut alors être réécrite :
Pour la constante d'équilibre K‡ , la mécanique statistique conduit à une expression dépendante de la température donnée par :
où
En combinant les nouvelles expressions pour k‡ et K‡,une nouvelle expression de la constante cinétique peut être écrite, donnée par :
Puisque ΔG = ΔH –TΔS, la constante cinétique peut être développée en ce qui est appelé l'équation d'Eyring :
L'expression de la constante cinétique TST peut être utilisée afin de calculer ΔG°‡, ΔH°‡, ΔS°‡ et parfois ΔV‡ (le volume d'activation) en utilisant les données cinétiques expérimentales.
Limites de la théorie de l'état de transition et développements ultérieurs
Limites de la théorie de l'état de transition
De manière générale, la TST a fourni aux chercheurs un outil conceptuel pour comprendre le déroulement des réactions chimiques. Bien que la théorie soit largement reconnue, elle a ses limitations. Ainsi, la théorie postule qu'une fois que la structure de transition descend la surface d'énergie potentielle, elle donne un produit (ou un ensemble de produits). Cependant, dans certaines réactions, cet état de transition peut traverser la surface d'énergie potentielle d'une manière telle qu'il conduise à une sélectivité non produite par la TST. C'est le cas par exemple de la réaction de décomposition thermique des diazaobicyclopentanes proposées par E.V. Anslyn et D.A. Dougherty.
La TST est aussi basée sur le postulat du comportement classique des noyaux atomiques[8]. De fait, on postule qu'à moins que les atomes ou les molécules se percutent avec assez d'énergie pour former les structures de transition, la réaction ne se produit pas. Cependant, selon la mécanique quantique, pour toute barrière d'énergie finie, il existe une possibilité que les particules puissent la traverser (effet tunnel). Si l'on considère les réactions, cela signifie qu'il existe une chance que les molécules puissent réagir même si elles ne se rencontrent pas avec assez d'énergie pour passer la barrière d'énergie[9]. Bien que cet effet soit a priori négligeable pour des réactions avec des énergies d'activation importantes, il devient beaucoup plus important pour des réactions avec des barrières relativement basses, la probabilité de « tunneling » croissant lorsque la hauteur de la barrière décroît.
La théorie de l'état de transition échoue à décrire certaines réactions à hautes températures. La théorie postule que le système réactionnel passera par le point-selle de plus basse énergie, dont le point le plus énergétique est appelé état de transition, sur la surface d'énergie potentielle. Si cette description est cohérente avec les réactions se produisant à des températures relativement basses, dans les hautes températures, les objets chimiques occupent des modes d'énergies vibrationnelles plus élevés. Leurs mouvements deviennent plus complexes et les collisions peuvent conduire à des états de transition très éloignés de ceux prédits par l'énergie d'état de transition. Cet éloignement à la TST peut être observé même dans l'échange simple entre l'hydrogène diatomique et un radical hydrogène[10].
Étant données ces limitations, de nombreuses alternatives ont été développées.
Théories de l'état de transition généralisées
Toute modification de la TST pour laquelle l'état de transition n'est pas nécessairement localisé au point selle, telles que la TST variationnelle microcanonique, la TST variationnelle canonique ou la TST variationnelle canonique améliorée, est référencée comme une théorie de l'état de transition généralisée.
- TST variationnelle microcanonique
En anglais Microcanonical Variational TST. Un des développements de la TST dans lequel la surface divisante est modifiée de façon à minimiser la cinétique pour une énergie fixée. Les expressions cinétiques obtenues par un traitement microcanonique peuvent être intégrées sur l'énergie, en prenant en compte la distribution statistique sur les états d'énergie, donnant ainsi les cinétiques canoniques (ou thermiques).
- TST variationnelle canonique
En anglais Canonical Variational TST. Développement de la TST dans laquelle le position de la surface divisante est modifiée afin de minimiser la constante cinétique à température donnée.
- TST variationnelle canonique améliorée
En anglais Improved Canonical Variational TST. Modification de la TST variationnelle canonique dans laquelle, pour des énergies en dessous d'une énergie seuil, la position de la surface divisante est celle de l'énergie seuil microcanonique. Cela force les contributions aux constantes cinétiques à être nulles si elles sont en deçà de l'énergie seuil. Une surface divisante de « compromis » est ensuite choisie de façon à minimiser les contributions à la constante cinétique des réactifs ayant les énergies les plus élevées.
Application de la TST : cas des réactions enzymatiques
Article principal : Cinétique enzymatique.Les enzymes catalysent des réactions chimiques avec des cinétiques stupéfiantes comparées à la chimie non catalysée dans les mêmes conditions réactionnelles. Chaque processus catalytique requiert un minimum de trois étapes, toutes se déroulant dans les quelques millisecondes qui caractérisent les réactions enzymatiques typiques. Selon la théorie de l'état de transition, la plus petite partie du cycle catalytique correspond à l'étape la plus importante, celle de l'état de transition. Les propositions initiales de théorie de la constante cinétique absolue pour des réactions chimiques définissaient l'état de transition comme une espèce distincte dans le déroulement de la réaction, espèce déterminant la constante cinétique absolue. Peu de temps après, Linus Pauling proposait que la puissante action catalytique des enzymes puisse être expliquée par une liaison forte et spécifique aux espèces d'état de transition[11]. La constante cinétique étant proportionnelle à la proportion de réactif dans le complexe d'état de transition, l'enzyme était considérée comme pouvant accroître la concentration d'espèces réactives.
Cette hypothèse a été formalisée par Wolfenden et collaborateurs au sein de l'Université de Caroline du Nord à Chapel Hill, qui supposèrent que l'accroissement cinétique imposé par les enzymes est proportionnel à l'affinité de l'enzyme pour la structure de l'état de transition par rapport au complexe de Michaelis-Menten[12]. Les enzymes accélérant les réactions par des facteurs de 1010-1015 par rapport aux réactions non catalysés, et les complexes de Michaelis-Menten ayant parfois des constantes de dissociation de l'ordre de 10−3-10−6 M, il fut proposé que les complexes d'état de transition sont liés avec des constantes de dissociation de 10-14 -10−23 M. Lorsque le substrat évolue du complexe de Michaelis-Menten au produit, la chimie se produit par des modifications enzymatiques dans la distribution électronique dans le substrat.
Les enzymes modifient la structure électronique par protonation, déprotonation, transfert électronique, distorsion géométrique, partition hydrophobe et interaction entre acides et bases de Lewis. Ces actions sont effectuées par des modifications séquentielles des charges protéiques et de substrat. Lorsqu'une combinaison de forces faibles individuelles s'exercent sur le substrat, la somme des énergies individuelles donne des forces capables de relocaliser les électrons liants afin de créer ou de défaire des liaisons. Des analogues ressemblant aux structures d'état de transition pourraient fournir les plus puissants inhibiteurs non-covalents connus, même si seule une petite fraction de l'énergie de l'état de transition est captée.
Toutes les transformations chimiques se produisent via une structure instable appelée état de transition, qui se situe en équilibre les structures chimiques des substrats et des produits. Les états de transitions pour des réactions chimiques sont supposés avoir des temps de vie proches de 10-13 secondes, de l'ordre de durée d'une seule vibration de liaison. Aucune méthode physique ou spectroscopique[réf. nécessaire] n'est disponible pour observer directement la structure d'un état de transition pour des réactions enzymatiques, alors que la structure de l'état de transition soit essentiel pour comprendre la catalyse enzymatique, les enzymes fonctionnant en minimisant l'énergie d'activation d'une transformation chimique.
Il est maintenant admis que les enzymes permettent la stabilisation des états de transition se trouvant entre les réactifs et les produits, et qu'elles devraient se lier fortement à tout inhibiteur ressemblant fortement à un tel état de transition. Les substrats et les produits participent souvent à de nombreuses réactions enzymatiques, alors que l'état de transition tends à être caractéristique d'une réaction enzymatique particulière, ce qui rend un inhibiteur spécifique d'une enzyme particulière. L'identification des nombreux inhibiteurs d'état de transition est un argument en faveur de l'hypothèse de stabilisation pour la catalyse enzymatique.
Il y a actuellement un grand nombre d'enzymes connues pour interagir avec des analogues de l'état de transition, la plupart étant conçues pour inhiber l'enzyme cible. On peut citer comme exemple la protéase de HIV-1, les racémases, les lactamases β, les métalloprotéinases, ou encore les cyclooxygénases.
- Cas de la purine nucléoside phosphorylase
La purine nucléoside phosphorylase (PNP) est une enzyme impliquée dans le catabolisme et le recyclage des nucléosides, et constitue une cible pour le développement de nouveaux agents thérapeutiques pour l'apoptose des lymphocytes T dans le cas de la leucémie et les maladies auto-immunes[13] L'inosine, la guanosine et la 2'-déoxyganosine sont les principaux substrats de cette enzyme (la figure 3 montre une réaction catalysée caractéristique de la PNP avec un substrat inosine).
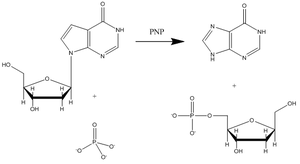 Figure 3 : phosphorolyse de l'inosine catalysée par la PNP.
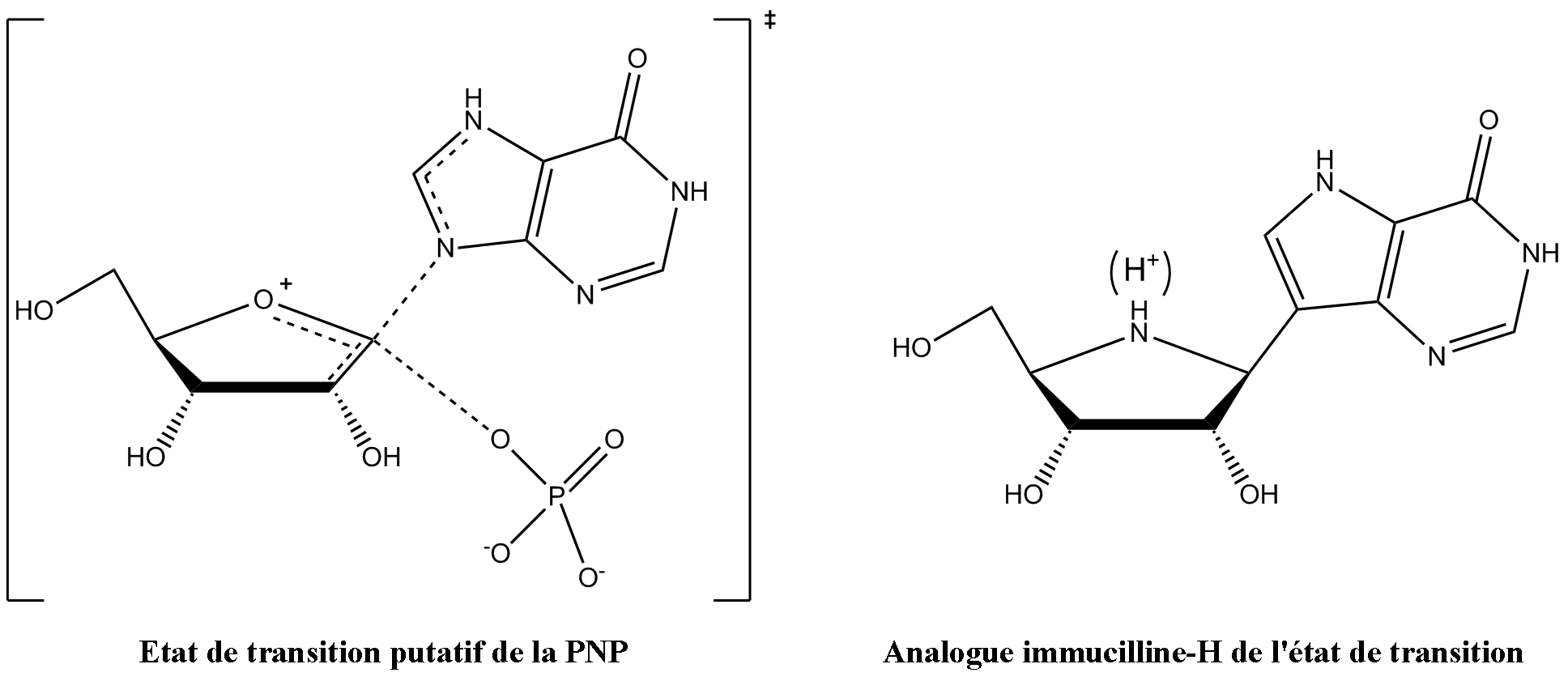
Figure 3 : phosphorolyse de l'inosine catalysée par la PNP.Vern Schramm et ses collaborateurs au sein de l'Albert Einstein College of Medicine ont déterminé la structure de l'état de transition de la PNP et l'ont utilisé afin de développer des analogues à liaisons fortes de l'état de transition afin d'inhiber cette enzyme[14]. L'inhibiteur immucilline H (forodésine) de la PNP est très proche structurellement de l'état de transition putatif (figure 4), avec des modifications nombreuses afin de rendre le composé plus stable que l'éphémère état de transition.
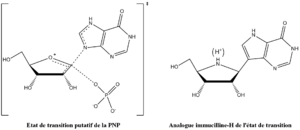 Figure 4 : état de transition dans la phosphorolyse de l'inosine, stabilisé par la PNP (à gauche), et immucilline H, de structure analogue (à droite).
Figure 4 : état de transition dans la phosphorolyse de l'inosine, stabilisé par la PNP (à gauche), et immucilline H, de structure analogue (à droite).La structure de l'état de transition de la phosphorolyse, stabilisée par la PNP, montre, pour la protonation, un pKa élevé pour la position N7 du cycle purine et sert comme donneur de liaison hydrogène avec l'oxygène de la chaine carbonyle latéral de l'asparagine 243, ce qui est imité dans l'immucilline H par utilisation de la 9-déazapurine en lieu et place de l'hypoxanthine. L'état de transition forme également un ion oxocarbénium dans le cycle de l'ose, qui est fourni par la partie iminoribitol qui possède une liaison ribosidique plus stable. L'ion phosphure n'est pas fortement impliqué dans la formation de liaison dans l'état de transition, et donc a été écarté de la conception de l'analogue de l'état de transition.
Les analogues de l'état de transition ont des propriétés d'inhibiteurs à liaison lente où dans la première étape l'inhibiteur se lie afin de former un complexe réversible EI, qui est suivi par une modification conformationnelle lente qui conduit à un complexe EI* très fortement lié.

 et
et 
Le KI a été déterminé déterminé par titrage de l'immucilline H et par mesure de son effet sur les vitesses initiales de la PNP vo, et sa valeur est de 41 nM. KI* a été calculé à partir des mêmes mesures cinétiques, mais au lieu d'utiliser les vitesses initiales, les vitesses vs de deuxième étape ont été utilisées, ce qui correspond aux cinétiques inhibées de l'état stable atteint après l'équilibre pour l'étape à initialisation lente lorsque tous les E ont formé des EI.
La stœchiométrie de la liaison de l'immucilline H indiquait qu'une molécule d'inhibiteur se lie à chaque trière de PNP, et une molécule était suffisante pour une inhibition enzymatique. Il avait été démontré auparavant que toute l'activité catalytique de la PNP est supportée par un seul site à la fois, de manière similaire à la protéine F1-ATP synthase. Il a été aussi relevé que la liaison d'analogues du substrat, du produit ou de l'état fondamental peut être réalisée sur les trois sites. Une preuve structurelle appuie l'hypothèse inhibitrice sur un tiers des sites pour cet analogue à l'état de transition, et tous les analogues de l'état fondamental montrent une occupation enzymatique totale.
La conception de l'immucilline H à partir d'une analyse de l'état de transition enzymatique illustre une puissante approche de développement d'inhibiteurs d'enzymes d'intérêt majeur pour l'industrie pharmacologique.
Notes et références
- Notes
- (en) International Union of Pure and Applied Chemistry (IUPAC). Transition State Theory. (consulté le 23 novembre 2008).
- (en) Truhlar, D. G.; Garrett, B. C.; Klippenstein, S. J., Current Status of Transition-State Theory, The Journal of Physical Chemistry 1996, 100, (31), 12771-12800.
- (en) Laidler, K.; King, C., Development of transition-state theory, The Journal of Physical Chemistry 1983, 87, (15), 2657
- (en) Laidler, K.; King, C., A lifetime of transition-state theory, The chemical intelligencer 1998, 4, (3), 39
- (en) Laidler, K. J., Theories of Chemical Reaction Rates (McGraw-Hill Series in Advanced Chemistry). 1969; p 234 pp.
- (en) Eric V. Anslyn and Dennis A. Dougherty. Transition State Theory and Related Topics. In Modern Physical Organic Chemistry University Science Books: 2006; pp 365-373
- (en) Laidler, K. J., Theories of Chemical Reaction Rates (McGraw-Hill Series in Advanced Chemistry). 1969; p 234 pp
- (en) Eyring, H.; Journal of Chemical Physics, 1935, 3, 107-115
- (en) Masel, R. Principles of Adsorption and Reactions on Solid Surfaces; Wiley, New York, 1996
- (en)Pineda, J. R.; Schwartz, S. D.; Philosophical Transactions of the Royal Society B 2006, 361, 1433-1438
- (en) Pauling, L.; American Scientist 1948, 36, 50-58.
- (en) Radzicka, A., Wolfenden, R. Science 1995, 267, 90-93.
- (en) Ringia, E.A.T.; Tyler, P.C.; Evans, G.B.; Furneaux, R.H.; Murkin, A.S.; Schramm, V.L., Transition State Analogue Discrimination by Related Purine Nucleoside Phosphorylases. Journal of American Chemical Society 2006, 128 (22), 7126-7127.
- (en) Miles R.W., Tyler P.C., Furneaux R.H., Bagdassarian C.K., Schramm V.L. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry. 1998, 37, (24), 8615-21
- Références
- (en) Laidler, K.; King, C., Development of transition-state theory. The Journal of physical chemistry 1983, 87, (15), 2657
- (en) Laidler, K., A lifetime of transition-state theory. The chemical intelligencer 1998, 4, (3), 39
- (en) Eric V. Anslyn, Dennis A. Doughtery., Transition State Theory and Related Topics. In Modern Physical Organic Chemistry University Science Books: 2006; pp 365–373
- (en) Schramm, V.L., Enzymatic Transition States and Transition State Analog Design. Annual Review of Biochemistry 1998, 67, 693-720
- (en) Schramm, V.L., Enzymatic Transition State Theory and Transition State Analogue Design. Journal of Biological Chemistry 2007, 282, (39), 28297-28300
- (en) Radzicka, A.; Woldenden, R., Transition State and Multisubstrate Analog Inhibitors. Methods in Enzymology 1995, 249, 284-312
- (en) Cleland, W.W., Isotope Effects: Determination of Enzyme Transition State Structure. Methods in Enzymology 1995, 249, 341-373
Voir aussi
- Principe de Curtin–Hammett
Lien externe
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Transition state theory » (voir la liste des auteurs)
Catégories :- Cinétique chimique
- Théorie chimique








![\mathrm{A} + \mathrm{B} \rightleftharpoons [\mathrm{AB}]^\ddagger \to \mathrm{P}](f/7af022438d43d800e23144645faf38e1.png)
![K^{\ddagger o} = \frac{[AB]^\ddagger}{[A][B]}](f/a3fcbd9a3fc1178f6954a71c386f8ccb.png)
![[\mathrm{AB}]^{\ddagger} = K^{\ddagger^o}[\mathrm{A}][\mathrm{B}]](7/b47be0cf7ca7618e31b09b3360ec8271.png)
![\frac{d[P]}{dt} = k^{\ddagger o}[\mathrm{AB}]^{\Dagger} = k^{\ddagger}K^{\Dagger }[A][B] = k[A][B]](f/77f5fe0d56143226afe44c13dafe3ffc.png)







Wikimedia Foundation. 2010.