- Lymphohistiocytose hémophagocytaire
-
Lymphohistiocytose hémophagocytaire
Classification et ressources externes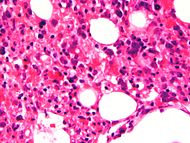
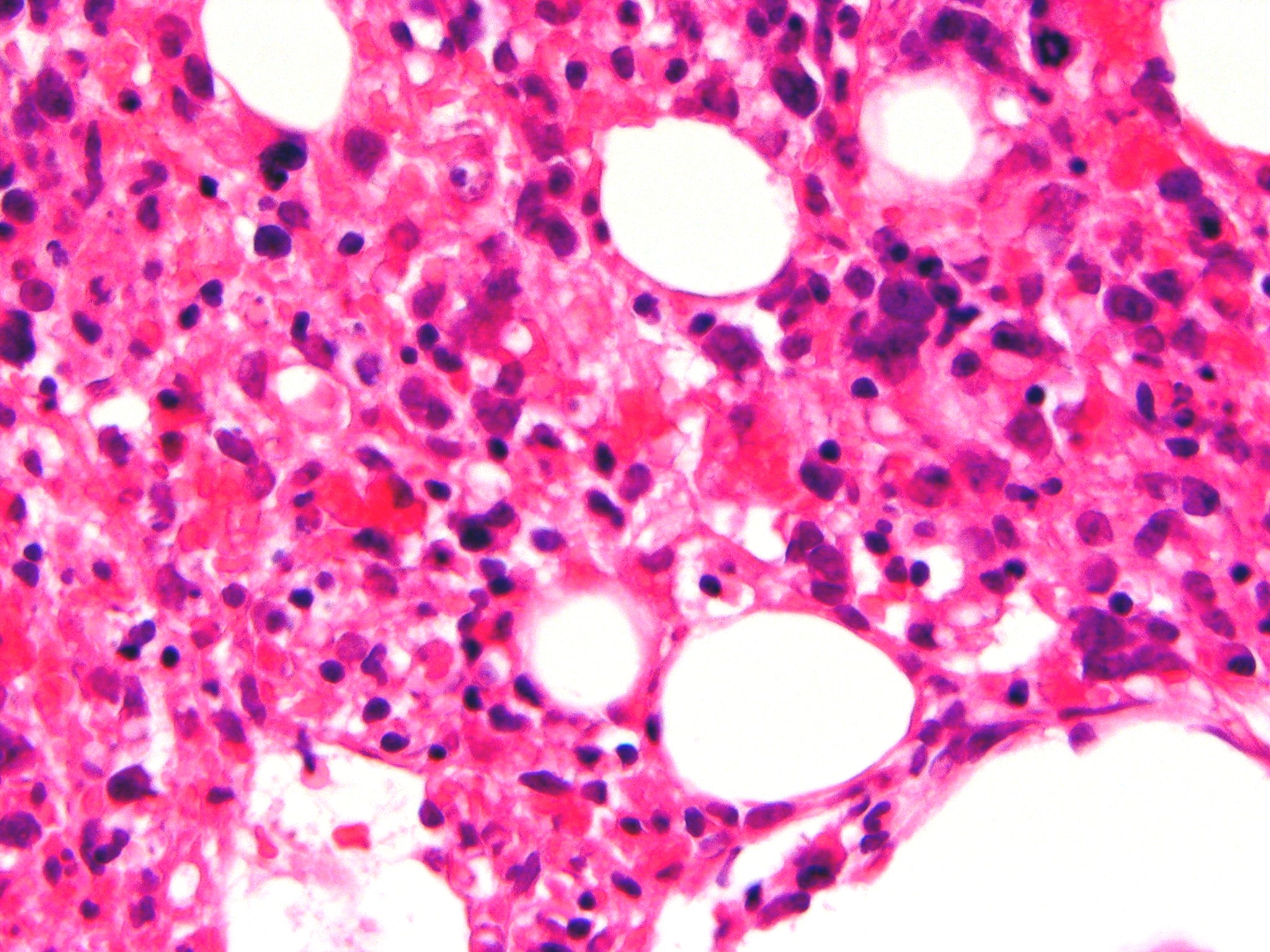
Moelle osseuse avec prolifération de macrophages contenant des globules rouges dans leurs cytoplasmes. CIM-10 D76.1 CIM-9 288.4 OMIM 267700 DiseasesDB 31418 eMedicine ped/745 MeSH D051359 La lymphohistiocytose hémophagocytaire appartient au groupe des histiocytoses non langerhansiennes et non malignes. Ce syndrome est marqué par la prolifération inappropriée de macrophages bénins activés, caractérisée par une hémophagocytose intense. Mais aussi, par un défaut de l’activité cytotoxique des lymphocytes NK et des lymphocytes T CD8+ ainsi que par une production de cytokines supérieure à la normale (hypercytokinémie). On distingue deux formes de lymphohistiocytose hémophagocytaire : la forme familiale ou LHF (lymphohistiocytose familiale) et les autres formes (associées à des infections virales, des maladies auto-immunes…). La lymphohistiocytose hémophagocytaire familiale (ou primaire) est une maladie héréditaire rare et rapidement fatale, à transmission autosomique récessive. Elle touche 1 naissance sur 50000. Elle apparaît généralement dès les premiers mois de la vie et le diagnostic peut s’avérer difficile.
Sommaire
Historique
En 1939, la réticulose médullaire histiocytaire a été mise en évidence, elle associe une altération de l’état général de l’organisme, une organomégalie et une pancytopénie périphérique. Les premiers cas de lymphohistiocytose hémophagocytaire familiale (LHF) ont été décrits par Farquhar et Claireaux en 1952. En 1966, Rappaport décrit une prolifération systémique d’histiocytes atypiques et regroupe ses observations sous le terme d’histiocytose maligne. En 1979, Risdall rapporte que le syndrome hémophagocytaire peut être aussi associé à une infection virale.
Tableau clinique
La maladie se manifeste par des signes biologiques et cliniques diverses :
- Les symptômes généraux évoluent très rapidement dès leur apparition, généralement une forte fièvre s’installe accompagnée de sueurs et de frissons.
- Une organomégalie, généralement une splénomégalie ou une hépatomégalie. Celles-ci sont dues à une infiltration par le contingent histiocytaire. Cependant dans les formes infantiles, l’organomégalie peut prendre un aspect pseudo tumorale.
- Des atteintes neurologiques comme l’ataxie, des crises convulsives, une irritabilité, encéphalopathie. Souvent ces troubles peuvent entrainer la mort des patients.
- Une atteinte cutanée : purpura, œdème, ictère le plus souvent, éruptions cutanées, érythème.
- Une atteinte pulmonaire : dans 20 à 30% des cas, il y a observation d’infiltrats pulmonaires sur les radios des poumons.
- Des signes digestifs peuvent démontrer la présence de la maladie : hémorragies digestives causée par une probable thrombopénie, des diarrhées.
- Une atteinte rénale
- Une atteinte cardiaque
Le diagnostic de la LH se fait sur la base de la présence de 5 au moins des symptômes suivants :
- Fièvre
- Splénomégalie
- Absence de malignité
- Cytopénies touchant au moins deux lignées hématopoïétiques :
- hémoglobine < 90g/L (enfants de moins de 4 semaines < 10g/L)
- plaquettes < 100 x 109 /L
- neutrophiles < 1.0 x 109 /L
- Triglycérides ≥ 265 mg/dL
- Fibrinogène ≥ 1.5 g/L
- Hémophagocytose dans la moelle osseuse ou dans la rate voire dans les ganglions lymphatiques
- Taux bas ou absence de l’activité des NK
- Ferritine ≥ 500 µg/L
- CD25 soluble ≥ 2400 U/mL
Dans les formes familiales de LH, on recherche bien sûr les mutations sur les gènes connus pour être associés à la maladie (diagnostic moléculaire).
Mécanismes moléculaires et physiopathologiques
La lymphohistiocytose est caractérisée par un défaut de l’activité cytotoxique des lymphocytes NK et lymphocytes TCD8+, une suractivation des macrophages et une hypercytokinémie. Ces défauts sont la conséquence de mutations de certains gènes caractéristiques des lymphohistiocytoses hémophagocytaires familiales. Ces mutations touchent des gènes codant des molécules essentielles au processus d’exocytose des granules lytiques et donc à la cytotoxicité.
- gène PRF1 : gène de la perforine (protéine cytolytique sécrétée par les lymphocytes T CD8+) localisée sur le chromosome 10q21-22 est muté chez 15 à 50% des patients présentant une LHF.
- gène UNC13D : gène qui code la protéine Munc13-4 localisé sur le chromosome 17q25 normalement responsable de l’arrimage des granules cytolytiques en préparation pour la fusion avec la membrane plasmique. Environ 15 à 30% des patients ayant une LHF présente cette mutation.
- gène STX11 : gène localisé sur le chromosome 6q24 codant la syntaxine 11 qui participe à la fusion des membranes vésiculaire et cellulaire. Cette mutation est présente chez environ 20% des patients d’origine turque présentant une LHF.
Le résultat de ces déficiences est double : mauvaise élimination des antigènes, donc hyperstimulation du système immunitaire avec une forte sécrétion de cytokines ; et défaut de régulation des macrophages par les cellules NK. En conséquence, il y a accumulation de lymphohistiocytes dans les organes et notamment dans les espaces périvasculaires et les méninges du cerveau. L’hypercytokinémie est responsable de la plupart des signes cliniques et biologiques observés de la LHF tout comme la variation d’interleukines en particulier IL-1 qui sont responsables de la fièvre. Un niveau élevé de TNF-α et d’interférons-γ (INF-γ) entrainent une intense fatigue, une sudation importante ainsi qu’une pancytopénie.
Traitements
Le traitement se détermine en fonction de la sévérité des symptômes et de la cause de la maladie, de manière à contrôler les symptômes. Pour les formes familiales, on donne un traitement immunosuppresseur en attendant une greffe de moelle osseuse. L’étoposide a été utilisé avec succès. Pour les formes secondaires, il faut surtout traiter le facteur déclenchant (infections notamment) et administrer un traitement immunosuppresseur également. Les drogues utilisées :
- Corticostéroïde
- Ciclosporine A
- Etoposide
- Injection d’immunoglobulines en intraveineuse
- Allogreffe de moelle osseuse
Bibliographie
- When T cells and macrophages do not talk: the hemophagocytic syndromes, Robert J. Arceci, Current Opinion in Hematology 2008, 15: 359-367
- Immunologie, Guy GOROCHOV et Thomas PAPO, Inter Med, 2000, 19: 177-186
- Lymphohistiocytose hémophagocytaire familiale
Wikimedia Foundation. 2010.