- Maladie De Horton
-
Maladie de Horton
 Pour les articles homonymes, voir Horton.
Pour les articles homonymes, voir Horton.Maladie de Horton
Classification et ressources externesCIM-10 M31.5 CIM-9 446.5 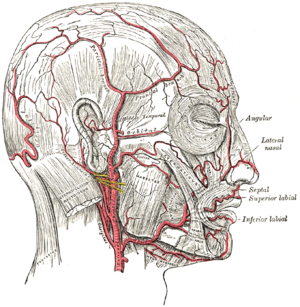 Les artères du visage
Les artères du visage
La maladie de Horton (ou artérite giganto-cellulaire) est une maladie inflammatoire des vaisseaux. Cette maladie touche particulièrement les sujets âgés. Elle est connue aussi sous le nom d'artérite temporale, du fait que cette artère est souvent affectée au cours de la maladie.
Cette maladie est d'incidence moyenne, le nombre de nouveaux cas estimé varie entre 6 et 22/100 000 par an[1].Sommaire
Historique et épidémiologie
Dès le Xe siècle, Ali Ibn Isa de Bagdad décrit l'association d’une cécité et d’une artérite temporale. Ensuite Hutchinson, en 1890, fait la première description clinique de l’artérite gigantocellulaire. Enfin en 1932, Bayard Taylor Horton et Magath font la description histopathologique de l’artérite temporale à cellules géantes à propos de deux cas.
La maladie de Horton ou artérite gigantocellulaire – c’est le terme retenu dans les publications internationales – est une artériopathie inflammatoire chronique du sujet âgé de plus de 50 ans. C’est la plus fréquente des vascularites chez les sujets caucasiens, son incidence est évaluée à une vingtaine de cas par an pour 100000 habitants de plus de 50 ans[2], elle est plus élevée dans les pays scandinaves[3]. L’âge moyen de survenue de cette maladie est 71 ans. L’incidence augmente avec l’âge : 90% des sujets atteints ont plus de 60 ans. Les sujets d’origine asiatique et africaine sont rarement atteints. Il existe une prédominance féminine marquée : deux femmes pour un homme.
Anatomopathologie
Sur le plan histopathologique, il s’agit d’une panartérite (atteinte de l'ensemble des artères) segmentaire et plurifocale à cellules géantes. La paroi des artères atteintes présente trois types de lésions associées ou non à la présence de cellules géantes :
- une infiltration par des lymphocytes, des monocytes et des polynucléaires neutrophiles au niveau de l’intima et de la média des artères atteintes,
- une destruction des fibres musculaires lisses de la média,
- une fragmentation de la limitante élastique interne.
La présence de l’infiltrat leucocytaire est constatée pour la majeure partie des vascularites. Par ailleurs une élévation du taux d’acide ribonucléique messager (ARNm) codant des cytokines (IL-2, IFN-gamma et IL-1-alpha) a été montrée à la fois dans des cas de maladie de Horton et dans des pseudopolyarthrites rhizoméliques (PPR). Malgré ces connaissances l’étiopathogénie de la maladie de Horton demeure inconnue.
Macroscopiquement la maladie de Horton atteint des artères de moyen et de gros calibre, principalement les branches de la carotide externe, avec une affinité particulière pour l’artère temporale superficielle.
Présentations cliniques
La forme complète et typique de la maladie de Horton comprend :
- Un syndrome d'altération de l'état général: une fièvre ou un fébricule au long cours, un amaigrissement et une fatigue (asthénie)
- des maux de têtes (céphalées) surtout au niveau des tempes avec une hypersensibilité du cuir chevelu (hyperesthésie)
- l'artère temporale peut sembler parfois dure et ne "bat" plus (abolition ou diminution du pouls temporal)
- Le symptôme le plus caractéristique est la claudication de la mâchoire. Le patient ne peut pas mastiquer très longtemps sans ressentir une douleur et être obligé de faire une pause dans son activité masticatoire.
Mais la maladie de Horton est une maladie qui touche tous les vaisseaux sanguins et les manifestations peuvent être très diverses : douleurs articulaires, atteintes des gros vaisseaux (carotide, aorte, artères iliaques), du cœur, des poumons, manifestations neurologiques ou psychiatriques, etc.
Elle est souvent associée à la PPR (pseudo-polyarthrite rhizomélique).
Hypothèses étiopathogéniques
RAHMAN et RAHMAN évoquent un découpage de la maladie de Horton en deux phases successives : une phase systémique (dominée par les signes généraux) qui précède une phase ophtalmique. Cette hypothèse est également défendue par une large étude rétrospective (portant sur 167 patients) : les patients diagnostiqués le plus tardivement après les premiers symptômes sont également ceux qui ont le plus fort taux d’atteintes ophtalmiques.
Une alternative à cette hypothèse consiste à penser que les différents modes de présentation clinique correspondent à différentes localisations d’artérite gigantocellulaire.
Enfin certains auteurs expliquent les différents modes de présentation par une potentielle diversité des étiologies, notamment auto-immunes et infectieuses.
Les signes céphaliques
Les signes cliniques « historiques » de la maladie de Horton sont donc céphaliques : céphalées, hyperesthésies du cuir chevelu (signe du peigne), épaississement douloureux des artères temporales, claudication de la mâchoire, diplopie, baisse d’acuité visuelle.
Selon les études, une atteinte oculaire unilatérale ou bilatérale est présente dans 14 à 70% des cas de la maladie de Horton. La complication grave la plus fréquente est en effet la cécité qui est liée à l’histoire naturelle de la maladie. Elle interviendrait dans 36% des cas : les études les plus anciennes parlent de 50% alors que la plus récente parle de 13%. Cette évolution décroissante du taux d’atteinte oculaire peut certainement être reliée à une meilleure prise en charge dans les études les plus récentes. Le diagnostic doit donc être fait le plus tôt possible.
Les atteintes extracraniales
Cependant cette définition historique de la maladie de Horton a été étoffée dès 1938 par les premières observations d’atteintes extracraniales. En 1946, Cooke et Al.[réf. nécessaire] n’hésitent pas à parler de « maladie vasculaire généralisée ». D’autres études autopsiques, réalisées dans les années 1960, vont argumenter un rapprochement nosologique entre la maladie de Horton et la pseudopolyarthrite rhizomélique. Il s’agirait en effet de deux entités répondant à une même définition anatomopathologique, à savoir une panartérite gigantocellulaire. L’hypothèse d’une nature systémique de l’artérite gigantocellulaire est actuellement la plus défendue. La différence de présentation clinique serait donc due à une diversité de localisation des atteintes vasculaires. De nombreuses localisations extracraniales rares ont été rapportées dans la littérature : utérus et ovaires, vaisseau thoraciques, artères coronaires, thyroïde, foie, intestin grêle, vésicule biliaire, rein, pancréas, œsophage, moelle osseuse, moelle épinière, nerfs et prostate.
Hiérarchisation sémiologique
Les signes cliniques les plus fréquemment retrouvés dans la maladie de Horton sont :
- la céphalée, signe clinique le plus fréquemment retrouvé, dans 60 à 90% des cas selon les auteurs ;
- une anomalie à la palpation des artères temporales (induration, nodule, diminution ou abolition du pouls) ;
- troubles visuels (diplopie, neuropathie optique ischémique antérieure ou postérieure, choroïdopathie ischémique, amaurose fugace, douleur oculaire, hallucinations) ;
- tableau de PPR (myalgies proximales, faiblesse musculaire, asthénie) ;
- signes généraux de l’inflammation (fièvre, anorexie, amaigrissement, asthénie, sueurs nocturnes) ;
- tableau d’ischémie subaiguë des membres supérieurs ;
- claudication de la mâchoire.
Aucun de ses signes n’a de valeur pathognomonique. De nombreux cas de maladie de Horton sont rapportés dans la littérature avec une symptomatologie non spécifique : anorexie, amaigrissement, asthénie, fébricule au long cours (≥ 3 mois), fièvre d’origine inexpliquée, syndrome inflammatoire inexpliqué, sueurs nocturnes, découverte fortuite d’une anémie.
Définition d’une fièvre d’origine indéterminée
Une fièvre d’origine indéterminée – ou fièvre d’origine inconnue – était définie de manière classique par une température corporelle supérieure à 38,3°C documentée à plusieurs reprises sur une durée minimale de 3 semaines et dont l’étiologie restait incertaine après trois jours d’investigations hospitalières correctement menées ou trois visites ambulatoires successives. Les trois étiologies les plus fréquentes des fièvres d’origine inexpliquées sont respectivement les infections, les néoplasies et les maladies inflammatoires chroniques non infectieuses] (maladies auto-immunes, vascularites).
Pseudo-polyarthrite rhizomélique et maladie de Horton
Il a été rapporté que certains patients souffrant d’une maladie de Horton avaient des signes cliniques évoquant une pseudo-polyarthrite rhizomélique. La maladie de Horton est associée dans environ 40% des cas à une pseudo-polyarthrite rhizomélique. Dès 1965 une étude autopsique retrouvait une artérite gigantocellulaire chez 5 patients sur 6 souffrant d’une pseudo-polyarthrite rhizomélique. Ces résultats ainsi que les apports récents de la tomographie à émission de positon montrent que la maladie de Horton n’a pas une localisation unique et font penser que la pseudo-polyarthrite rhizomélique est en fait une forme de vascularite proche de la maladie de Horton, peut-être une variante de localisation.
Atteinte aortique (dite aortite de Horton)
La maladie de Horton, malgré sa définition « historique » ne se cantonne pas aux vaisseaux de l’extrémité céphalique. Elle peut atteindre l’ensemble des vaisseaux de moyen et gros calibre, notamment les vaisseaux thoraciques de gros calibre. L’incidence de ces atteintes dans les séries autopsiques, ces atteintes est fréquentes, entre 50 et 70%, ce qui est corroboré, in vivo, par les études par tomographie à émission de positons[4]. Elles sont toutefois de reconnaissance difficile.
L’augmentation de l’incidence des anévrismes et dissections aortiques chez les patients atteints de Horton est l'une des complications gravissimes de la maladie : on estime le risque relatif à 17,3. Ces complications sont parfois mortelles.
La surmortalité liée à la maladie de Horton n’a pas été montrée de manière claire. Deux études rétrospectives sont arrivées à des conclusions différentes. L’une sur une période de suivi de 12 ans n’a pas montré de surmortalité, l’autre s’étend sur une période presque 50 ans et montre que les patients souffrant d’une atteinte des gros vaisseaux ont une surmortalité par rapport à ceux qui ont une atteinte purement céphalique. Dans la série rétrospective de LIE et Al., 12 des 35 patients souffrant d’une aortite de Horton sont décédés subitement de rupture d’anévrisme ou de dissection aortique. AGARD et Al. ont mené une étude scanographique de l’aorte sur une série prospective de 22 patients atteints de MH comparés à 22 témoins appariés pour l’âge et le sexe. Ils ont montré une prévalence accrue d’anomalies morphologiques : épaississements pariétaux, ectasies et anévrismes de l’aorte.
Les autres artères du corps humain peuvent être touchées : artères sous-clavières, artères à destinée céphalique en dehors des artères temporales, artères des membres inférieurs. Une série de 72 patients, suivis à la Mayo Clinic, ayant un diagnostic histologique d’artérite gigantocellulaire, a montré que les atteintes extracraniales de cette pathologie étaient par ordre de fréquence décroissante : l’aorte (35/72), les vaisseaux de la tête, du cou et des membres supérieurs (24/72) et enfin les vaisseaux des membres inférieurs (13/72).
Complications
La complication grave la plus fréquente est la cécité. Elle peut survenir très brusquement et est définitive. Elle est présente dans un peu moins de 15% des cas[5].
Diagnostic
Le diagnostic est fondé sur trois caractéristiques cliniques (âge supérieur à 50 ans, céphalées d'apparition récente, induration et diminution de la pulsatilité de l'artère temporale) et deux caractéristiques paracliniques (VS élevée (95%) supérieure à 50, inflammation granulomateuse à la biopsie de l'artère temporale (80%))[6].
L'échographie de l'artère temporale est parfois utile lorsque la suspicion de diagnostic est faible. Les signes échographiques à rechercher sont le signe du halo, la sténose ou l'occlusion de l'artère.
Les critères diagnostiques de l’American College of Rheumatology
En 1990, l’ACR, réuni en conférence de consensus, a énoncé des critères diagnostiques pour certaines vascularites dont la maladie de Horton. Trois des critères suivants permettent de faire le diagnostic d’une maladie de Horton :
- âge supérieur ou égal à 50 ans,
- céphalée localisée de début récent,
- artère temporale indurée ou diminution/abolition du pouls temporal,
- vitesse de sédimentation (VS) supérieure à 50 mm à la première heure,
- biopsie artérielle positive montrant une infiltration par des mononucléaires ou une inflammation granulomateuse avec ou sans cellules géantes.
Leur sensibilité est de 93,5% et leur spécificité de 91,2%.
Place des examens complémentaires dans le diagnostic et le suivi
Biologie
Les présentations frustes posent des problèmes en matière d’outils diagnostiques. En effet, il n’existe pas de test biologique spécifique. Cependant, on peut mettre en évidence biologiquement le syndrome inflammatoire par la VS et la CRP, la CRP ayant l’avantage de ne pas varier avec l’âge. La conjonction d’une accélération de la VS et d’une augmentation de la CRP a une spécificité de 97% pour le diagnostic de MH. Par ailleurs on note la présence fréquente d’une thrombocytose. Les anticorps anti-cardiolipine sont présents chez plus de 50% des patients. Enfin le taux d’IL-6 est élevé, notamment chez les patients qui présentent des symptômes non spécifiques. D'autres marqueurs augmentent parfois aussi : le fibrinogène, la 5' nucléotidase.
Biopsie de l’Artère Temporale (BAT) et critères ACR
Le diagnostic positif est histologique : il est obtenu par la biopsie de l’artère temporale. Actuellement cette biopsie peut être réalisée dans des conditions tout à fait acceptables, de plus elle ne laisse aucune séquelle. Deux problèmes diagnostiques se posent principalement.
Premièrement la BAT est positive dans seulement 70% des maladies de Horton dont le tableau clinique est typique. Quelques études rapportent une sensibilité supérieure, entre 80 et 90%. BLOCKMANS parle de 16% de faux négatifs. Une biopsie controlatérale après une première biopsie négative peut être réalisée mais elle est positive dans 3 à 10% des cas seulement. Il faut souligner que la biopsie semble ne pas perdre de sensibilité si elle est pratiquée dans les 5 premiers jours après instauration de la corticothérapie. Après 15 jours de corticothérapie, la sensibilité s’effondre.
Deuxièmement, les présentations cliniques non spécifiques rendent le diagnostic encore plus difficile. La BAT est positive dans seulement 25% des maladies de Horton à localisations extracraniales. Dans une autre étude 42% des patients ayant une MH avec atteinte des gros vaisseaux avaient une BAT négative.
La biopsie de l’artère temporale ne serait positive que dans 0 à 41% des tableaux de PPR, une méta-analyse parle de 10% et c’est sans doute la valeur à retenir.
Ce sont ces tableaux atypiques de maladie de Horton regroupant des signes non spécifiques pour lesquels le diagnostic est difficile. Quelles investigations complémentaires sont utiles et dans quel ordre les réaliser si cette biopsie n’apporte pas le diagnostic ?
La maladie de Horton peut être définie de la manière suivante si la BAT est négative :
- présence de trois critères ACR et absence d’autre diagnostic susceptible d’expliquer la symptomatologie dans les 6 derniers mois, diagnostic confirmé par deux expertises médicales distinctes (16) ;
- une autre définition semble se dessiner actuellement : présence de trois critères ACR et présence de signes typiques de vascularite sur des examens d’imagerie.
L’imagerie dans la MH
L'échographie transthoracique, l'échographie transœsophagienne, l'échographie abdominale, la scintigraphie au Gallium 67, la tomodensitométrie, l'angioscanner, l'imagerie par résonance magnétique, l'angioIRM (=ARM), la tomographie par émission de positons au 18FDG ont été proposées dans cette démarche diagnostique.
L’angiographie, l’angioTDM et plus récemment l’ARM sont les examens « gold standard » utilisés pour le diagnostic des vascularites des gros vaisseaux, mais les lésions mises en évidence ne sont pas spécifiques et peuvent correspondre à des lésions banales d’origine athéromateuse chez le sujet âgé. L’avantage principal de l’angiographie conventionnelle est sa résolution jusqu’à 500 μm. Mais l’angiographie ne permet pas de faire le diagnostic à un stade précoce.
La scintigraphie au Gallium n’a pas donné de bons résultats.
L'échographie doppler a été étudiée dans les années 1980 mais sa spécificité inférieure à 80% ne lui a pas permis de rivaliser avec la biopsie de l'artère temporale. L’échographie doppler est utilisée par certaines équipes pour rechercher de manière systématique une anomalie vasculaire sur les gros troncs thoraciques (sténose, dissection, occlusion).
Actuellement le doppler couleur couplé est l'un des meilleurs examens pour le diagnostic : il retrouve un halo hypoéchogène autour du vaisseaux, une hypoéchogénicité de l'intima du vaisseau touché par l'inflammation, une sténose ou une occlusion. Nous nous intéresserons à la valeur diagnostique du halo qui est le signe le plus précoce. La sensibilité du doppler couleur couplé est de 40 à 100% par rapport à l'histologie, selon treize études, avec une médiane à 86%. La sensibilité comparée au diagnostic clinique est comprise entre 35 et 86 % avec une médiane à 70%). Sa spécificité en comparaison de l’histologie est de 93% en médiane sur les mêmes études. Elle est de 97% comparée au diagnostic clinique. Cette spécificité est bonne mais il s’agit d’un examen opérateur dépendant : il faut donc un opérateur entraîné à cette indication particulière. Sa résolution est de 0,1 à 0,2 mm. Par contre il est impossible de couvrir par cet examen l’ensemble du corps humain notamment les gros vaisseaux thoraciques.
L'intérêt principal de l'échographie est d'orienter le choix du site de biopsie pour potentialiser la positivité de cette biopsie : une Bbiopsie de l'artère temporale réalisée après repérage échographique retrouve un infiltrat lymphocytaire chez 100% des sujets et des cellules géantes chez 75% des sujets. AGARD et Al. la réservent aux cas où la biopsie paraît hasardeuse, notamment en cas d’abolition du pouls temporal. La revue Prescrire a proposé tout récemment, dans une mise au point, le recours à l’échographie dans les cas où la suspicion clinique est faible, afin d’éviter un geste invasif au patient. La définition de ces situations ne repose pas pour le moment sur des critères standardisés. De plus il est intéressant de préciser que les cas de suspicion clinique faible sont également les cas où l’atteinte des vaisseaux céphaliques est la moins fréquente.
Diagnostic différentiel
Les douleurs temporales peuvent aussi provenir d'une contracture des fibres musculaires du faisceau antérieur / moyen / postérieur du muscle temporal : c'est souvent le cas dans la dysfonction crânio-mandibulaire (syn. "malocclusion dentaire", syndrome temporo-mandibulaire, SADAM, STM, etc.). Dans la dysfonction manducatrice, les douleurs temporales ont toujours un côté préférentiel, à caractère dominant sur l'autre versant, et s'accompagnent d'autre foyers myalgiques homolatéraux, d'une déglutition salivaire dysfonctionnelle (interpositions linguales, déglutition infantile, "cheveu sur la langue", etc.), de malpositions dentaires, etc.
Traitement
Les anti-inflammatoires stéroïdiens sont la base du traitement de la Maladie de Horton.
Le traitement, à mettre en route le plus rapidement possible, est la corticothérapie car il s’agit d’une urgence ophtalmologique. Ce traitement à la cortisone commence avec des doses assez importantes qui seront diminuées petit à petit. Le traitement dure en général plus de dix-huit mois. Il est souhaitable de commencer ce traitement avant même la biopsie de l'artère temporale.
La dose initiale de Prednisone retenue varie entre 0,7 et 1 mg/Kg/jour selon les publications. Cette dose est adaptable à la baisse en cas de syndrome inflammatoire peu prononcé, ou à la hausse en fonction des répercussions oculaires, vasculaires ou neurologiques. Les européens semblent préconiser des doses plus faibles que les nord-américains [réf. nécessaire].
La décroissance progressive des doses est actuellement guidée par les marqueurs biologiques de l’inflammation : Protéine C réactive (CRP) et VS. La CRP a comme principal avantage de ne pas varier avec l’âge, mais elle a l’inconvénient de refléter une inflammation aiguë. Elle diminue donc très rapidement après le début de la corticothérapie. La VS est accélérée par l’âge mais reflète une inflammation sur une durée plus longue. De plus il existe une formule pour corréler la VS à l’âge. La VS est mieux corrélée à la fibrinémie que la CRP. Il n’y a pas de consensus pour déterminer lequel de ces deux marqueurs est le plus fiable. Les européens semblent utiliser plus volontiers la CRP tandis que les nord-américains utilisent davantage la VS.
La récidive est fréquente (près d'un tiers des cas)[7], surtout au cours de la première année.
Utilisée depuis les années 1950, la corticothérapie reste le traitement conventionnel malgré l’avènement des traitements immunosuppresseurs, notamment dans les cas de corticorésistance. En cas d'échec de la corticothérapie, le méthotrexate peut être essayé. Le recours au Méthotrexate est très controversé [réf. nécessaire]. Par contre, l’utilisation d’un antiagrégant plaquettaire semble diminuer le risque de survenue de complications vasculaires ou ophtalmologiques. Enfin, en cas d’atteinte oculaire ou vasculaire grave, certains auteurs préconisent des bolus de Méthylprednisolone de 250 mg/jour sur 3 jours.
Notes et références
- ↑ ORPHANET - Maladies rares - Médicaments orphelins
- ↑ Salvarani C, Gabriel SE, O'Fallon WM, Hunder GG. Epidemiology of polymyalgia rheumatica in Olmsted County, Minnesota, 1970-1991. Arthritis Rheum. 1995;38:369-73
- ↑ Gran JT, Myklebust G. The incidence of polymyalgia rheumatica and temporal arteritis in the county of Aust Agder, south Norway: a prospective study 1987-94. J Rheumatol. 1997;24:1739-43
- ↑ Blockmans D, de Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients, Arthritis Rheum, 2006;55:131-7
- ↑ González-Gay MA, García-Porrúa C, Llorca J, Hajeer AH, Brañas F, Dababneh A, et al. Visual manifestations of giant cell arteritis. Trends and clinical spectrum in 161 patients. Medicine (Baltimore). 2000;79:283-92
- ↑ critères proposés par l'American College of Rheumatologie
- ↑ Bengtsson BA, Malmvall BE. Prognosis of giant cell arteritis including temporal arteritis and polymyalgia rheumatica. A follow-up study on ninety patients treated with corticosteroids, Acta Med Scand, 1981;209:337-45
Liens externes
- Artérite à cellules géantes, fiche résumé sur Orphanet, et Maladie de Horton et Pseudopolyarthrite rhizomélique, dossier pdf complet sur Orphanet.
 Portail de la médecine
Portail de la médecine
Catégories : Vascularite | Maladie en rhumatologie | Maladie vasculaire et oeil | Céphalée | Urgence médicale | Maladie en gériatrie

Wikimedia Foundation. 2010.