- Lysosomes
-
Lysosome
 Les principaux organites d'une cellule. Les lysosomes sont légendés en haut à gauche
Les principaux organites d'une cellule. Les lysosomes sont légendés en haut à gauche
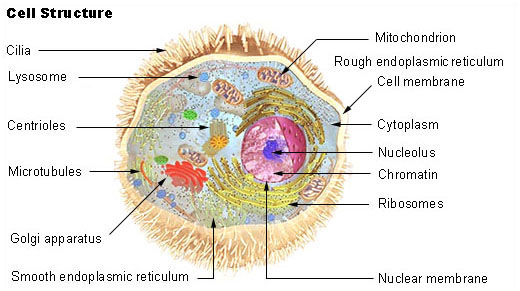
 Schéma d'une cellule animale typique. Organites: (1) nucleole (2) noyau (3) ribosome (4) vésicule (5) réticulum endoplasmique granuleux (REG) (6) Appareil de Golgi (7) Cytosquelette (8) Réticulum endoplasmique lisse (9) mitochondries (10) vacuole (11) cytoplasme (12) lysosome (13) centrioles
Schéma d'une cellule animale typique. Organites: (1) nucleole (2) noyau (3) ribosome (4) vésicule (5) réticulum endoplasmique granuleux (REG) (6) Appareil de Golgi (7) Cytosquelette (8) Réticulum endoplasmique lisse (9) mitochondries (10) vacuole (11) cytoplasme (12) lysosome (13) centriolesLes lysosomes sont des organites cellulaires de 0,2 à 0,5 microns présents dans le cytosol de toutes les cellules eucaryotes animales, à l'exception des hématies (« globules rouges »)[1]. Ils ont pour fonction d'effectuer la digestion intra-cellulaire (ou extra-cellulaire via exocytose dans le cas des chondroblastes, ostéoclastes et macrophages) grâce à trois types d'enzymes : des lipases, des protéases et des osidases. La membrane lysosomale contient des protéines de transport, des pompes à protons pour l'hydrogène (protons H+) et des canaux ioniques spécifique aux ions chlorures Cl-. Ces pompes à protons et ces canaux ioniques permettent le maintien à l'intérieur des lysosomes d'un pH compris entre 3,5 et 5, indispensable au fonctionnement des hydrolases acides qu'ils contiennent[2]. Les lysosomes sont formés dans l'appareil de Golgi.
Christian de Duve, en 1957, distinguait les lysosomes primaires n'ayant pas encore rencontré de matériel à digérer, et les lysosomes secondaires.
Actuellement la distinction porte sur la source du matériel digéré. Trois voies sont ainsi considérées :
- Des produits d'endocytose, contenus dans des endosomes fusionnent avec les vésicules de lysosome, chargées d'hydrolases, pour former les endolysosomes.
- Les organites cellulaires détruits s'entourent d'une membrane provenant du réticulum endoplasmique. Ceci forme un autophagosome, qui, par fusion avec un endolysosome ou un lysosome aboutirait à la formation d'un autophagolysosome. Les autophagolysosomes assurent le mécanisme d'autophagie cellulaire.
- Dans les cellules phagocytaires, les phagosomes (vésicules contenant des déchets ou une bactérie) sont transformés en phagolysosome par association avec un lysosome ou un endolysosome.
L'activité lysosomale est très importante chez le têtard dont la queue disparaît à l'état adulte. Le lysosome fut décrit et nommé pour la première fois en 1955 par Christian de Duve. L'anomalie du fonctionnement enzymatique d'une des enzymes contenues dans le lysosome est responsable de maladies lysosomales. Cela donne par exemple des maladies de surcharge lipidique (génétiques). Il peut y avoir formation de corps résiduels. Ce sont des vacuoles provenant de phagolysosomes ou d'autophagolysosomes dans lesquels persistent des résidus non digérés par les enzymes lysosomiales. Ils peuvent être de deux types : les figures myéliniques, et la lipofushine, qui résulte de l'oxydation enzymatique des lipides (surtout dans les vieux neurones).
Sommaire
Enzymes du lysosome: les hydrolases acides
Les lysosomes contiennent des enzymes digestives (hydrolases acides) pour digérer les macromolécules. Pour fonctionner correctement, les enzymes digestives requièrent l'environnement acide du lysosome. Pour cette raison, si des hydrolases acides devaient fuir vers le cytosol, leur danger potentiel pour la cellule serait réduit, car elles ne seraient pas à leur pH optimum. Toutes ces enzymes sont produites par le réticulum endoplasmique, et transportées et traitées par l'appareil de Golgi. Chaque hydrolase acide est ensuite ciblée vers un lysosome. Le lysosome lui-même est apparemment protégé de la digestion par ses structures tridimensionnelles internes uniques qui préviennent une action enzymatique[3]
Quelques enzymes importantes dans les lysosomes sont :
- Lipase, qui dégradent les lipides en acides gras.
- Glycoside hydrolases, qui dégradent les hydrate de carbone (les sucres),
- Protéases, qui dégradent les protéines en peptides, dégradés ensuite par peptidase en tripeptides, dipeptides, puis acides aminés.
- Nucléases, qui dégradent les acides nucléiques en nucléosides
Il y a donc dans le lysosome tous le matériel nécessaire à la dégradation d'une cellule.
Fonctions
- Hétérophagique : décomposition des nutriments.
- Autophagique : recyclage des éléments cellulaires abîmés, retraitement des substances toxiques produites par la cellule.
- Rôle dans les remaniements tissulaires (ex : rein → suppression progressive des ébauches grâce aux lysosomes).
- Rôle de détoxication : stockage des selles biliaires nocives pour la cellule.
- Rôle de régulation de la sécrétion hormonale de la cellule.
Maladies lysosomales ou Maladies lysosomiales
Il y a un bon nombre de maladies causées par un dysfonctionnement des lysosomes ou d'une de leurs protéines digestives, par exemple la maladie de Tay-Sachs, ou la maladie de Pompe. Elles sont dues à une protéine digestive manquante ou défectueuse, ce qui entraîne une accumulation de substrats dans la cellule, et de là un métabolisme cellulaire modifié. Globalement, les maladies lysosomales peuvent être classifiées comme mucopolysaccharidoses, gangliosidoses, trouble du stockage des lipides, glycoprotéinoses, mucolipidoses, ou leucodystrophies.
Après dégradation de certains produits par le lysosome il subsiste des résidus qui ne peuvent plus être dégradés mais s'accumulent dans le lysosome. Cela pourrait expliquer la courte durée de vie des cellules spécialisées dans la phagocytose comme les globules blancs (1 à 2 jours). Toutes les cellules sont capables de phagocyter et elles le feront par exemple lors d'apoptose de cellule voisine pour "nettoyer". Si pour les globules blancs l'accumulation de résidus n'est pas grave, elle l'est beaucoup plus pour les cellules qui ne se divisent plus comme les neurones, on pourrait donc déterminer l'âge d'un être par l'état de ses lysosomes neuronaux.
Liens externes
- (fr) Qu'est-ce qu'une maladie lysosomale ? Association Vaincre les Maladies Lysosomales
- (be) {http://www.maladies-lysosomales.be
Notes et références
- ↑ En effet, les cellules limitées par une paroi extracellulaire comme les cellules végétales ou les cellules constitutives des mycètes sont incapables d'endocytose et sont dépourvues de lysosomes.
- ↑ Le pH du cytosol est neutre (environ 7,2).
- ↑ Campbell, Neil A. and Reece, Jane B. (2002). Biology 6th ed. Benjamin Cummings. San Francisco. (ISBN 0805366245)
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire Portail de la médecine
Portail de la médecine
Catégorie : Organite

Wikimedia Foundation. 2010.