- Paralysie périodique thyrotoxique
-
Paralysie périodique thyrotoxique (PPT)
Classification et ressources externes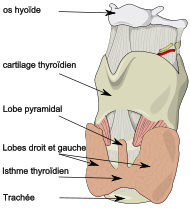
Localisation de la glande thyroïde dans le cou CIM-10 G72.3 CIM-9 359.3 OMIM 188580 DiseasesDB 29122 MedlinePlus 000319 eMedicine article/1171678 MeSH D020514 GeneReviews Hypokalemic Periodic Paralysis La paralysie périodique thyrotoxique (PPT) est une maladie génétique qui se traduit par des accès temporaires de paralysie. Elle est due à une hyperactivité de la glande thyroïde et fut identifiée la première fois par Carl Westphal en 1885. Elle touche généralement les individus de sexe masculin d'origine asiatique entre 20 et 50 ans. Elle se manifeste par la survenue brutale d'une faiblesse musculaire et/ou d'une paralysie sans atteinte sensitive chez des personnes hyperthyroïdiennes (ayant une hyperactivité de la glande thyroïde). Une hypokaliémie (baisse du taux de potassium dans le sang) est habituellement constatée au moment des crises. La crise peut conduire exceptionnellement à la mort si la faiblesse des muscles respiratoires entraîne une insuffisance respiratoire ou si l'hypokaliémie entraîne un trouble sévère du rythme cardiaque[1],[2]. Non traitées, ces crises ont généralement un caractère récurrent[1].
La maladie est liée à des mutations génétiques des gènes codant pour certains des canaux ioniques permettant le transport des électrolytes (sodium et potassium) à travers les membranes cellulaires. Les principales mutations portent sur le gène CACNA1S qui code pour la protéine formant la sous-unité Cav1.1 d'un canal calcique des muscles squelettiques[2] et le gène KCNJ18 qui code pour la protéine d'un canal potassique rectifiant entrant (Kir2.6)[3] ; la maladie est donc classée comme une canalopathie[3]. L'anomalie est considérée être liée à une entrée rapide du potassium à l'intérieur des cellules lors d'une hyperthyroxinémie (concentration anormalement haute d'hormone thyroïdienne dans le sang), généralement avec un facteur déclenchant supplémentaire. Le traitement de l'hypokaliémie, suivi de la correction de l'hyperthyroïdie, conduit à la disparition complète des crises.
La PPT est une des causes de paralysie périodique[4].
Sommaire
Historique
 Carl Friedrich Otto Westphal
Carl Friedrich Otto Westphal
Après plusieurs signalements de cas au XVIIIe et XIXe siècles, la paralysie périodique thyrotoxique a d'abord été décrite en détail par le neurologue allemand Carl Friedrich Otto Westphal (1833-1890)[5],[6]. En 1926, le médecin japonais Tetsushiro Shinosaki, de Fukuoka, observa un taux élevé de maladies de la thyroïde chez les Japonais atteints périodiquement de crises de paralysie[7],[8]. Le premier rapport en langue anglaise, en 1931, provient de Dunlap et Kepler, les médecins de la Mayo Clinic qui ont décrit une crise caractéristique chez un patient souffrant de la maladie de Basedow[2],[8]. En 1937, la paralysie périodique thyrotoxique est reliée à une hypokaliémie, qui répond au traitement associant glucose et insuline, un traitement aggravant habituellement l'hypokaliémie[9],[10]. Ce phénomène a été utilisé comme un test de diagnostic[10].
En 1974, il a été découvert que le propranolol pouvait prévenir les attaques[11]. La notion de canalopathies et le lien avec les mutations de canaux ioniques spécifiques a émergé à la fin du XXe siècle[1],[3],[4].
Symptômes
Les crises commencent le plus souvent par des douleurs musculaires, des crampes et des raideurs[12] suivies par une faiblesse et une paralysie qui tend à se développer rapidement, le plus souvent aux premières heures de la matinée ou tard le soir ; la faiblesse est généralement symétrique[12] (qui touche les deux moitiés du corps de façon analogue). Les muscles proximaux des membres (i. e. ceux des cuisses et des bras) sont plus touchés que les autres et la faiblesse a tendance à commencer dans les cuisses avant de se répandre dans les bras. Les muscles de la bouche et de la gorge, des yeux et de la respiration ne sont généralement pas touchés mais, parfois, une faiblesse des muscles respiratoires peut entraîner une insuffisance respiratoire potentiellement mortelle. Ces crises cessent généralement en 2 à 72 heures, même en l'absence de traitement[1],[2],[12]. À l'examen neurologique lors d'une crise, une faiblesse flasque des membres est observée, associée à une diminution des réflexes, mais le système sensitif[1],[12] de même que l'état mental[12] ne sont pas affectés.
Les crises peuvent être déclenchées par un effort physique, la consommation d'alcool ou d'aliments riches en glucides ou en sel. Cela peut expliquer pourquoi les attaques sont plus fréquentes en été, quand les gens boivent plus de boissons sucrées et font davantage d'exercice. Les crises liées aux efforts ont tendance à se produire au cours de la période de repos qui suit immédiatement l'exercice physique. La reprise d'un effort peut être recommandée pour faire avorter une crise survenant après un exercice physique[1].
Des symptômes d'hyperactivité thyroïdienne peuvent être observés sur les sujets, tels que perte de poids, tachycardie, tremblements et sueurs[1],[2], mais une fois sur deux, il n'y a aucun de ces symptômes[12]. Le type le plus commun d'hyperthyroïdie, la maladie de Basedow, peut en outre provoquer des problèmes oculaires (ophtalmopathie de Basedow) et des modifications de la peau des jambes (myxoedème prétibial)[13]. Toute hyperthyroïdie peut aussi provoquer une faiblesse musculaire (myopathie thyrotoxique) qui a tendance à être permanente et non pas épisodique[12].
Étiologies
Génétique
Les mutations dans les gènes qui codent pour la sous-unité α1 des canaux calciques de type-L (Cav1.1) associées à la PPT ont été décrites chez des individus du Sud de la Chine. Les mutations sont situées dans une partie du gène différente de celles décrites dans la paralysie périodique. Dans la PPT, les mutations décrites sont des polymorphismes nucléotidiques simples situés dans l'élément de réponse aux hormones thyroïdiennes, ce qui implique que la transcription du gène et la production de protéines de canaux ioniques peuvent être modifiées par une augmentation du taux d'hormones. Des mutations portant sur les gènes codant pour des canaux potassiques voltage-dépendants Kv3.4 et Canal sodium voltage-dépendant Na41.4 ont été également rapportées[1].
Parmi les personnes ayant une PPT, 33 % ont des mutations sur le gène KCNJ18, gène codant pour la protéine Kir2.6, d'un canal potassique rectifiant entrant. La régulation de l'expression de ce gène est aussi contrôlée par les récepteurs des hormones thyroïdiennes[3].
Certains types d'antigènes des leucocytes humains (HLA) comme B46, DR9, DQB1 * 0303, A2, Bw22, AW19, B17 et DRW8, sont trouvés plus fréquemment dans la PPT. Un lien entre la PPT et des types particuliers de HLA, qui jouent un rôle central dans la réponse immunitaire, pourrait impliquer une étiologie du système immunitaire à la maladie, mais on ne sait pas si cela provoque directement la PPT ou s'il accroît la susceptibilité à la maladie de Basedow, qui est une maladie auto-immune connue[1].
Maladie de la thyroïde
La forme la plus commune de maladie de la thyroïde associée à la PPT est la maladie de Basedow due à une réaction auto-immune qui conduit à une surproduction d'hormones thyroïdiennes[13]. La PPT a également été décrite chez les personnes atteintes d'autres troubles de la thyroïde telles qu'une thyroïdite, un goitre nodulaire toxique, un adénome toxique, un adénome thyréotrope, une ingestion excessive de thyroxine ou d'iode[1] et une hyperthyroïdie induite par l'amiodarone[2].
Mécanisme
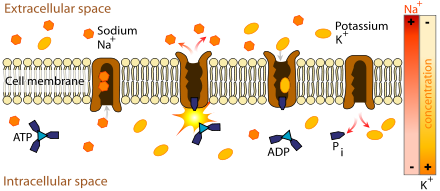 La Na+/K+-ATPase maintient constant le gradient de concentration sodium/potassium entre milieux intra et extracellulaires, en utilisant l'ATP comme source énergétique.
La Na+/K+-ATPase maintient constant le gradient de concentration sodium/potassium entre milieux intra et extracellulaires, en utilisant l'ATP comme source énergétique.La faiblesse musculaire et le risque accru d'arythmie cardiaque de la PPT sont dus à une baisse très forte de la kaliémie par rapport aux valeurs normales. Cette baisse n'est pas due à une perte de potassium mais à une augmentation de l'activité de la pompe Na+/K+-ATPase (l'enzyme qui fait pénétrer le potassium dans les cellules et rejette le sodium dans le plasma en proportion 2/3) augmentant la concentration de potassium dans les cellules et appauvrissant sa concentration sanguine. Dans d'autres types d'anomalies de concentration plasmatique du potassium, l'équilibre acido-basique est généralement perturbé, avec souvent soit une alcalose métabolique soit une acidose métabolique. Dans la PPT, ces perturbations sont généralement absentes. L'hypokaliémie entraîne une hyperpolarisation des cellules musculaires, ce qui rend la jonction neuromusculaire moins sensible à l'influx nerveux normal et conduit à une diminution de la contractilité musculaire[1].
Les raisons précises de l'augmentation de l'activité de la pompe Na+/K+-ATPase due aux anomalies génétiques décrites auparavant sont inconnues. Il est possible que l'enzyme devient plus actif en raison de l'augmentation des concentrations plasmatiques des hormones thyroïdiennes. L'hyperthyroïdie augmente la concentration sanguine en catécholamines (comme l'adrénaline), augmentant l'activité de la pompe Na+/K+-ATPase[12]. L'activité enzymatique est ensuite encore augmentée par d'autres causes. Par exemple, l'augmentation de l'apport en glucides conduit à une augmentation de l'insulinémie, qui est activatrice de la pompe Na+/K+-ATPase. Lorsque la cause est supprimée, le phénomène s'arrête[1]. Il a été postulé que la testostérone augmenterait l'activité de la Na+/K+-ATPase, ce qui expliquerait pourquoi les hommes sont plus exposés à la PPT alors que les maladies de la thyroïde sont plus fréquentes chez les femmes[2].
La PPT est considérée comme un modèle pour d'autres maladies analogues, connues sous le nom de « canalopathies » qui sont liées à des mutations touchant les canaux ioniques. La majorité de ces maladies se manifestent par des crises épisodiques[3].
Diagnostic
Une hypokaliémie est fréquente pendant les crises avec généralement des concentrations en potassium inférieures à 3,0 mmol/l. La magnésémie et la phosphatémie sont souvent diminuées. La concentration en créatine kinase (CPK) est trop élevée dans deux tiers des cas, ce qui est généralement dû à un certain degré de lésions musculaires ; des élévations critiques évocatrices d'une rhabdomyolyse (destruction sévère des tissus musculaires) sont rares[1],[2].
Un électrocardiogramme (ECG) peut montrer une tachycardie en raison de l'hyperthyroïdie, des anomalies comme une arythmie cardiaque (fibrillation auriculaire), une tachycardie ventriculaire et des changements de conduction dus à une hypokaliémie (présence d'ondes U, élargissement de QRS, allongement de QT et aplatissement de l'onde T)[2]. Un électromyogramme (EMG) montre des changements similaires à ceux rencontrés dans les myopathies (maladies du muscle), avec une amplitude réduite des potentiels d'action musculaire[4]. Ces signes disparaissent lorsque le traitement a commencé[1].
La PPT se distingue des autres formes de paralysie périodique (en particulier la paralysie périodique hypokaliémique familiale) par des dosages thyroïdiens anormaux. Ces dosages sont normaux dans les autres formes alors que dans la thyrotoxicose les niveaux de thyroxine et de triiodothyronine sont élevés, avec une baisse résultante de la production de TSH par l'adénohypophyse[1],[13]. Diverses autres recherches sont habituellement effectuées pour distinguer les différentes causes d'hyperthyroïdie[13].
Traitement
 Le propranolol diminue rapidement les symptômes de l'hyperthyroïdie.
Le propranolol diminue rapidement les symptômes de l'hyperthyroïdie.À la phase aiguë de la crise, l'administration de potassium permet en général d'améliorer rapidement la faiblesse musculaire et de prévenir les complications. Toutefois, il est prudent de rappeler que la quantité totale de potassium dans l'organisme n'a pas diminué et il est possible qu'avec le traitement les valeurs normales de la kaliémie soient dépassées (« hyperkaliémie de rebond »). Il faut poser une perfusion lente de chlorure de potassium avant que tout autre traitement ne soit commencé[1].
Les effets de l'hormone thyroïdienne en excès répondent généralement bien à l'administration d'un bêta-bloquant non sélectif tel que le propranolol (car la plupart des symptômes sont liés à des niveaux accrus d'adrénaline et à son effet sur les récepteurs β-adrénergiques). En outre, d'autres crises peuvent être prévenues en évitant les facteurs déclenchants connus tels que la consommation excessive de sel et de glucides jusqu'à ce que la maladie de la thyroïde soit correctement traitée[1].
Le traitement de la maladie de la thyroïde conduit généralement à la résolution des attaques de paralysie. Selon la nature de la maladie, le traitement peut consister en la prise de médicaments thyréostatiques (médicaments qui stabilisent la production d'hormones thyroïdiennes), l'iode radioactif, et parfois la thyroïdectomie[1],[2].
Épidémiologie
La PPT survient surtout chez les sujets masculins chinois, japonais, vietnamiens, philippins, coréens[1] et thaïlandais[3] avec des taux beaucoup plus faibles chez les personnes d'autres fonds génétique[1]. Chez les personnes chinoises et japonaises souffrant d'hyperthyroïdie, 1,8 à 1,9 % font des crises de PPT. Ceci contraste avec les habitants d'Amérique du Nord, où des études ont rapporté un taux de 0,1 à 0,2 %[1],[2]. Les Amérindiens, qui partagent une partie de leur patrimoine génétique avec les Asiatiques du Sud-Est, se sont révélés également avoir un risque accru[1].
L'âge moyen de survenue est de 20 à 40 ans. Les causes de cette pénétrance très majoritairement masculine, avec des taux allant de 17 à 70 fois celui des femmes suivant les auteurs[14] sont inconnues, alors que l'hyperactivité thyroïdienne est beaucoup plus fréquente chez les femmes[1],[2].
Références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Thyrotoxic periodic paralysis » (voir la liste des auteurs)
- (en) A.W. Kung, « Clinical review: Thyrotoxic periodic paralysis: a diagnostic challenge », dans J. Clin. Endocrinol. Metab., vol. 91, no 7, juillet 2006, p. 2490–2495 [texte intégral, lien PMID, lien DOI]
- (en) P. Pothiwala, S.N.Levine, « Analytic review: thyrotoxic periodic paralysis: a review », dans J. Intensive Care Med., vol. 25, no 2, 2010, p. 71–77 [lien PMID, lien DOI]
- (en) D.P. Ryan, L.J. Ptácek, « Episodic neurological channelopathies », dans Neuron, vol. 68, no 2, octobre 2010, p. 282–292 [lien PMID, lien DOI]
- (en) B. Fontaine, « Periodic paralysis », dans Adv. Genet., vol. 63, 2008, p. 3–23 [lien PMID, lien DOI]
- (de) CF Westphal, « Über einen merkwürdigen Fall von periodischer Lähmung aller vier Extremitäten mit gleichzeitigem Erlöschen der elektrischen Erregbarkeit während der Lähmung », dans Berl. Klin. Wochenschr., vol. 22, 1885, p. 489–91 et 509–11
- (en)D. Sternberg, N. Tabti, B. Hainque et B. Fontaine, « Hypokalemic Periodic Paralysis », 28-04-2009. Consulté le 23 décembre 2010 PMID 20301512
- (de) T. Shinosaki, « Klinische Studien über die periodische Extremitätenlähmung », dans Z. Gesamt. Neurol. Psychiatr., vol. 100, no 1, 1926, p. 564–611 [lien DOI]
- (en) H. Dunlap, K. Kepler, « A syndrome resembling familial periodic paralysis occurring in the course of exophthalmic goiter », dans Endocrinology, vol. 15, no 6, 1931, p. 541–546 [lien DOI]
- (en) R.S. Aitken, E.N. Allott, L.I. Castleden, M. Walker, « Observations on a case of familial periodic paralysis », dans Clin. Sci., vol. 3, 1937, p. 47–57
- (en) A.J. McFadzean, R. Yeung, « Periodic paralysis complicating thyrotoxicosis in Chinese », dans Br. Med. J., vol. 1, no 5538, février 1967, p. 451–455 [lien PMID, lien DOI]
- (en) R.T. Yeung, T.F. Tse, « Thyrotoxic periodic paralysis. Effect of propranolol », dans Am. J. Med., vol. 57, no 4, octobre 1974, p. 584–590 [lien PMID, lien DOI]
- (en) S.H. Lin, « Thyrotoxic periodic paralysis », dans Mayo Clin. Proc., vol. 80, no 1, janvier 2005, p. 99–105 [texte intégral [PDF], lien PMID, lien DOI]
- (en) A.P. Weetman, « Graves' disease », dans N. Engl. J. Med., vol. 343, no 17, octobre 2000, p. 1236–1248 [lien PMID, lien DOI]
- Paralysie périodique thyrotoxique, Orphanet

La version du 20 février 2011 de cet article a été reconnue comme « bon article », c'est-à-dire qu'elle répond à des critères de qualité concernant le style, la clarté, la pertinence, la citation des sources et l'illustration.



Wikimedia Foundation. 2010.