- Tetralogie de Fallot
-
Tétralogie de Fallot
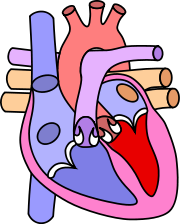 Cœur normal
Cœur normal
 Les deux anomalies de la Tétralogie de Fallot
Les deux anomalies de la Tétralogie de Fallot La description classique de cette cardiopathie (A : sténose, B : dextroposition de l'aorte, C : communication inter-ventriculaire, D : hypertrophie)
La description classique de cette cardiopathie (A : sténose, B : dextroposition de l'aorte, C : communication inter-ventriculaire, D : hypertrophie)La tétralogie de Fallot est la plus fréquente des cardiopathies congénitales cyanogènes. Elle représente près de 8% de l'ensemble des cardiopathies congénitales. Son nom est associé à Étienne-Louis Arthur Fallot qui l'a décrite en 1888.
Aujourd'hui, le diagnostic de cette malformation est possible avant même la naissance grâce à l'échographie fœtale. Dans la grande majorité des cas, le traitement, toujours chirurgical, est possible dès la première année de vie et consiste d'emblée en une « réparation complète » de la malformation. Le résultat de cette chirurgie s'apparente souvent à une guérison complète autorisant une vie normale.
Sommaire
Historique
La première description de la maladie en a été faite en 1671[1]. Étienne-Louis Arthur Fallot publie sa propre description en 1888[2].
Historiquement, en raison même de sa fréquence, il est probable que la plupart des enfants atteints de « la maladie bleue » décrits par nos grand-parents ou parents présentaient en fait une tétralogie de Fallot. C'est la première cardiopathie congénitale cyanogène pour laquelle a été proposée une intervention chirurgicale (1944), non pas curative mais palliative, c'est-à-dire améliorant les symptômes sans traiter réellement la malformation. Il s'agissait de l'anastomose de Blalock-Taussig. Dans les années 1950, il n'était pas rare de voir dans les journaux nationaux des souscriptions organisées pour permettre à des enfants « bleus » de traverser l'Atlantique (en bateau à l'époque) pour aller se faire opérer aux États-Unis d'une anastomose de Blalock-Taussig. La fin des années 1950, voit apparaître les premières tentatives de cures réparatrices.
Épidémiologie
La tétralogie de Fallot représenterait environ 10% des cardiopathies congénitales[3]. Son incidence est proche de 4 pour 10 000 naissances, soit 200 à 220 nouveaux cas en France chaque année (sur 783500 naissances répertoriées par l'INSEE en 2007). C'est la cardiopathie congénitale cyanogène la plus fréquente chez l'enfant (la transposition des gros vaisseaux étant la plus fréquente à la naissance, mais avec une certaines mortalité dans les premières semaines de vie). Avant la chirurgie, l'évolution en était presque toujours fatale avant l'âge de 30 ans.
Définition
Comme son nom l'indique, la tétralogie de Fallot comporte quatre anomalies, telles que Fallot, anatomo-pathologiste, avait pu les observer lors d'autopsies. En fait, deux de ces signes suffisent à expliquer la physiopathologie de cette malformation, les deux autres n'étant que des conséquences. Les deux anomalies principales sont :
1. La sténose (rétrécissement) de la voie d'éjection du ventricule droit. Elle peut siéger sur l'infundibulum pulmonaire (partie du ventricule droit située sous les valves pulmonaires), l'anneau pulmonaire et le tronc de l'artère pulmonaire (qui précède la bifurcation en deux artères pulmonaires). Ce rétrécissement peut être localisé ou prédominer sur l'une des ces trois parties dans les formes mineures de la malformation. A l'opposé, elle peut être sévère et s'étendre sur l'artère pulmonaire, y compris sur l'origine des branches droite et/ou gauche de l'artère pulmonaire. Le pronostic et les possibilités thérapeutiques dépendent surtout de cette anomalie Lésion A.
2. La présence d'une communication entre le cœur droit et le cœur gauche à travers la cloison séparant les deux ventricules (septum interventriculaire) (communication inter-ventriculaire). Cette communication siège dans la portion de la cloison appelée « septum conal », située à la partie supérieure du septum interventriculaire, proche de l'origine des gros vaisseaux et en particulier de l'aorte. Cette communication résulte d'un mauvais alignement entre l'aorte et la portion musculaire du septum interventriculaire.
Les deux autres anomalies décrites par Fallot ne sont que la conséquence des premières :
3. L'hypertrophie (épaississement) du ventricule droit est secondaire à l'augmentation de pression provoquée par la sténose pulmonaire. Cette hypertrophie est relativement modérée et en tout cas moindre que celle qui peut être observée en cas de sténose pulmonaire sévère isolée.
4. La dextroposition de l'aorte, c'est-à-dire son déplacement partiel au-dessus du ventricule droit n'est que la conséquence du défaut d'alignement évoqué à l'origine de la communication interventriculaire. Historiquement d'ailleurs, Fallot l'avait décrit car, lors de ses études anatomiques, il pouvait voir l'aorte directement au-dessus de la communication depuis l'intérieur du ventricule droit.
La trilogie de Fallot ne doit pas être confondue avec la tétralogie de Fallot. C'est le nom, de moins en moins usité, donné autrefois à l'association d'une sténose de l'artère pulmonaire et d'une communication entre les deux oreillettes.
Mécanisme
Dans le cœur normal:
- Les cœurs droit et gauche sont séparés, sans communication entre les deux systèmes.
- Les pressions à l'intérieur du cœur droit sont inférieures à celles régnant dans le cœur gauche.
- Le cœur droit pompe du sang pauvre en oxygène vers les poumons. Le cœur gauche pompe du sang riche en oxygène vers les organes.
L'association d'un rétrécissement de l'artère pulmonaire et d'une communication inter-ventriculaire provoque :
- Dans les cavités droites, une augmentation des pressions qui deviennent égales ou supérieures à celles des cavités gauches ;
- un passage de sang anormal, déterminé par les différences de pression, c'est-à-dire dans ce cas, du ventricule droit vers le ventricule gauche. Il s'agit d'un shunt droit-gauche.
Le sang éjecté dans l'aorte par le ventricule gauche contient en partie du sang oxygéné et en partie du sang pauvre en oxygène provenant du cœur droit. Ce sang appauvri en oxygène explique la cyanose.
Un shunt droit-gauche est caractéristique des cardiopathies cyanogènes dont fait partie la tétralogie de Fallot.
Diagnostic
Anténatal
Aspects échographiques avant la naissance
Le diagnostic de cette cardiopathie est possible avant la naissance par échographie, à partir de 22 SA (semaines d'aménorrhée) voire avant. En France, c'est le mode diagnostique dans 70 à 80% des cas (et jusqu'à 100% quand l'examen est fait par un(e) échographiste de référence)[4]. Le plus souvent, il s'agit d'une découverte 'de novo', sans antécédent familial particulier.
L'incidence la plus souvent pratiquée lors des échographies anténatales, dite « coupe des quatre cavités » est normale. Seule l'étude des gros vaisseaux issus du cœur et de leur connexion avec les cavités cardiaques permettra de mettre en évidence :
- Un signe d'appel : La dextroposition de l'aorte qui apparait « à cheval sur le septum interventriculaire » et non en continuité avec celui-ci comme normalement. Cette malposition de l'aorte, plus antérieure que normalement est responsable de la persistance d'une communication inter-ventriculaire dite « par malalignement » située immédiatement sous la naissance de l'aorte (CIV sous-aortique).
- Un signe diagnostique : L'hypoplasie de la voie pulmonaire.
Cette hypoplasie, ou défaut de développement, associe une diminution du diamètre de l'anneau pulmonaire (où s'attachent les valves pulmonaires) à une diminution du tronc de l'artère pulmonaire et peut s'étendre à l'origine des branches pulmonaires droite et gauche. En amont, la portion du ventricule droit d'où nait le tronc pulmonaire (appelé « chambre de chasse » ou « infundibulum pulmonaire ») apparait elle-même comme rétrécie en raison de la saillie anormale de bandelettes musculaires dans la cavité. Cette sténose infundibulaire est habituellement peu marquée chez le fœtus mais présente une tendance naturelle à l'aggravation progressive dans les mois suivant la naissance. Habituellement, il s'y associe une malformation des valves pulmonaires responsable d'une sténose valvulaire pulmonaire. L'importance de ce rétrécissement ne pourra être correctement évalué qu'après la naissance mais, à la différence de la sténose infundibulaire, il est habituellement de sévérité moyenne et a tendance à rester stable dans le temps.
Lorsque le diagnostic de Tétralogie de Fallot est évoqué, il convient de confier le fœtus à un échographiste de référence, si possible secondé par un cardio-pédiatre pour:
- Préciser la gravité anatomique. Il serait plus exact de parler « des » Tétralogies de Fallot que « de la » Tétralogie de Fallot. Tous les intermédiaires sont possibles entre une CIV sous-aortique isolée avec voie pulmonaire normale et la forme extrême que constitue l' « Atrésie pulmonaire à septum ouvert ». Le pronostic, les difficultés chirurgicales et la qualité du résultat opératoire dépendront bien évidemment de la sévérité de l'anomalie.
- Rechercher d'autres anomalies cardiaques associées, en particulier:
- La présence d'autres CIV qui compliqueraient la cure chirurgicale,
- La présence d'anomalie(s) des valves auriculo-ventriculaires entrant dans le cadre du « Canal atrio-ventriculaire » et dont la constatation ferait particulièrement craindre l'association à une Trisomie 21 ou syndrome de Down.
- Procéder à un examen morphologique complet du fœtus à la recherche d'anomalies extra-cardiaques, la Tétralogie de Fallot pouvant être observée dans de nombreux syndromes polymalformatifs (voir plus bas).
Les diagnostics différentiels :
La Tétralogie de Fallot fait partie d'un groupe de malformations cardiaques issus d'une anomalie commune: un défaut d'évolution de la région cono-truncale de l'ébauche cardiaque. Les autres anomalies cono-troncales qui partagent avec la tétralogie de Fallot le même signe d'appel échographique (« aorte à cheval sur une CIV ») sont :- La Communication inter-ventriculaire sous-aortique isolée. La voie pulmonaire est de calibre normale. Le traitement après la naissance sera celui d'une « simple » CIV avec le bon pronostic qui s'y rattache;
- L'Atrésie pulmonaire à septum ouvert (APSO) où au contraire, l'anneau pulmonaire est atrétique, imperméable, et le tronc pulmonaire tellement hypoplasique qu'il n'est habituellement pas visible en échographie. De la voie pulmonaire ne reste que la bifurcation et les artères pulmonaires, alimentées à contre-courant par la canal artériel.
- Le Tronc artériel commun, malformation où une seule artère (le « tronc commun ») donne à la fois l'aorte et les artères pulmonaires.
Prise en charge de la grossesse
Le diagnostic de Tétralogie de Fallot incite à proposer une amniocentèse pour étude du caryotype foetal, les deux principales anomalies chromosomiques à éliminer étant la microdélétion 22q11 et la trisomie 21.
Un suivi échographique de la grossesse permet d'apprécier la croissance de la voie pulmonaire et d'affiner le pronostic.
L'accouchement (par voie basse sauf indication obstétricale à une césarienne) sera programmé de préférence dans une maternité disposant d'un service de réanimation pédiatrique (en France, maternité dite « de niveau III ») afin de surveiller étroitement le nouveau-né au moment de la fermeture du canal artériel. En cas de déclenchement prématuré du travail, le fait d'accoucher dans une autre maternité « n'est pas une catastrophe ». Certes, il faudra envisager un transfert rapide du nouveau-né mais celui ci ne nécessite habituellement pas de traitement spécifique dès la naissance.
Prise en charge du nouveau-né
Dans sa forme habituelle, la Tétralogie de Fallot est bien supportée à la naissance et ne demande pas de traitement immédiat. Il est cependant utile d'hospitaliser le nouveau-né dans une unité de réanimation pédiatrique pour surveillance. Celle-ci portera essentiellement sur la qualité de l'oxygénation de l'enfant, appréciée par la mesure continue (monitorage) de la saturation en oxygène du sang circulant grâce à un petit capteur placé sur la peau ou à l'extrémité d'un doigt. A défaut, la saturation en oxygène peut être appréciée de façon discontinue, par prélèvement sanguin dans une artère ou au niveau des capillaires. Cette surveillance est maintenue jusqu'à la fermeture du canal artériel c'est-à-dire jusqu'à ce que la circulation du nouveau-né se soit établie selon le modèle définitif.
Dans les formes sévères, les nouveau-nés peuvent d'emblée présenter une cyanose - couleur bleutée des téguments et des muqueuses - plus ou moins importante, traduisant une désaturation en oxygène importante. Afin d'éviter qu'elle ne s'aggrave, il pourra être indiqué de maintenir artificiellement ouvert le canal artériel grâce à la perfusion de Prostaglandines puis de réaliser rapidement une anastomose de Blalock-Taussig, intervention qui consiste à créer l'équivalent d'un canal artériel permanent en interposant un tube entre une artère sous-clavière et une Artère pulmonaire (voir plus bas).
Postnatal
Clinique
Nouveau-né, nourrisson
À la naissance, le nouveau-né est le plus souvent bien coloré, rose, sans gêne particulière. L'attention est cependant attirée par la présence d'un souffle cardiaque, systolique, perçu à l'auscultation très tôt, dès la salle d'accouchement. Un souffle d'apparition aussi précoce doit faire pratiquer une échocardiographie en urgence.
La date d'apparition de la cyanose, signe classique de la malformation dépend de sa forme anatomique. Habituellement, elle est retardée de plusieurs semaines et n'est observée au début que lors des efforts ou des colères. Au repos, elle reste très modérée et ne peut être détectée que par un observateur averti. Elle s'accentue peu à peu, s'accompagnant progressivement de fatigue gênant l'enfant dans ses acquisitions motrices.
Habituellement, il n'y a pas de manifestation d'insuffisance cardiaque, l'enfant s'alimente correctement et sa croissance staturo-pondérale est normale, au moins dans les premiers mois de vie.
- La complication principale : le malaise anoxique.
Un malaise anoxique est lié à une augmentation brutale de la sténose infundibulaire qui interrompt plus ou moins complètement le passage du sang dans les artères pulmonaires (et donc son oxygénation) et accentue le passage de sang « bleu » (désaturé en oxygène) vers l'aorte au travers de la CIV. Il s'en suit une désaturation brutale et profonde du sang dans l'aorte et un manque d'oxygène (« hypoxie ») dans les principaux organes dont en particulier le cerveau. Cette hypoxie cérébrale se traduit par divers troubles neurologiques : l'enfant, plus ou moins geignard, le teint grisâtre, devient hypotonique (« poupée de chiffon »), semble absent et présente éventuellement des mouvements convulsifs. Le malaise se lève le plus souvent à ce moment, laissant l'enfant abattu. Si le malaise se poursuivait, l'enfant présenterait alors de véritables convulsions avec un risque de décès.
C'est donc une complication grave et au moindre doute de malaise, une consultation hospitalière s'impose en urgence (pas dans une semaine, pas demain, tout de suite !).Petit enfant
Helen Taussig avait décrit sous le terme de « squatting » une attitude particulière que pouvaient prendre spontanément les enfants porteurs d'une Tétralogie de Fallot, en particulier après l'effort.[5]. Ce comportement peut apparaitre vers l'âge de 6 mois et consiste à adopter une position couchée « en chien de fusil », les cuisses repliées sur l'abdomen. Plus tard, quand l'enfant peut se tenir debout, il s'agit d'un accroupissement. Ces positions permettent effectivement une amélioration plus rapide de la désaturation artérielle en oxygène secondaire à un effort. il existe une explication physiopathologique à ce type de comportement. L'accroupissement coude les veines fémorales et diminue ainsi le retour de sang vers le ventricule droit qui peut mieux l'éjecter dans la voie pulmonaire. Il coude aussi les artères fémorales et augmente les résistances artérielles systémiques, ce qui contribue à diminuer le passage de sang du ventricule droit vers l'aorte. Ces deux phénomènes concourent à une meilleure oxygénation sanguine.
En cas de malaise anoxique survenant à domicile, replier les cuisses de l'enfant sur son abdomen pourrait être un réflexe utile de la part des parents (en attendant l'hospitalisation).
Grand enfant, adulte
Il est de plus en plus rare de rencontrer une Tétralogie de Fallot non opérée après l'âge de deux ans. Soit il s'agit d'une forme particulière, soit le patient vit dans un pays en voie de développement. Le tableau est alors celui d'une cyanose plus ou moins profonde, s'accompagnant d'une fatigue et d'une dyspnée limitant les possibilités d'effort, par manque d'oxygène et non par insuffisance cardiaque.
- Les complications peuvent être de trois ordres :
- malaise anoxique comme pour le nourrisson ;
- complications liées à la polyglobulie : Fatigue, maux de tête, vertiges, thrombose veineuse ou artérielle (et alors dans un organe comme la rate, le rein ou surtout le cerveau à l'origine d'un accident vasculaire cérébral). La thrombose peut se constituer sur place ou être secondaire à la migration d'un caillot veineux. Normalement un tel caillot vient se bloquer dans les poumons, donnant une embolie pulmonaire. Dans la Tétralogie de Fallot et en raison du shunt droit-gauche au travers de la CIV, ce caillot peut passer dans l'aorte et aller se bloquer dans un organe (« embolie paradoxale »).
- complications infectieuses : D'une part, la Tétralogie de Fallot comporte un risque majeur d'endocardite, d'autre part, comme pour un caillot sanguin (« thrombus cruorique »), un thrombus infecté d'origine veineuse peut migrer par la grande circulation et infecter un organe par voie artérielle.
Ainsi, avant l'ère chirurgicale, on pouvait dire qu'un enfant porteur d'une tétralogie de Fallot qui n'était pas décédé d'un malaise anoxique mourrait plus tard d'un abcès cérébral ou d'une endocardite.
Examens complémentaires
L'échographie cardiaque est maintenant le principal (et souvent le seul) examen complémentaire fait pour affirmer le diagnostic et préciser la forme anatomique.
D'autres examens peuvent néanmoins s'avérer très utiles :
- L'électrocardiogramme (ECG) :
En montrant des signes d'hypertrophie ventriculaire droite (HVD) de type « adaptation » ou « égalité de pressions », l'ECG peut aider à rectifier un diagnostic erroné de simple CIV chez le nouveau-né. Ainsi, la présence d'une onde T positive en V1 (critère simple mais non unique d'HVD) chez un nouveau-né présentant un tableau clinique de CIV bien tolérée doit conduire à remettre en cause ce diagnostic et faire soupçonner la présence d'une Tétralogie de Fallot.
- La radiographie thoracique :
Elle apporte aussi, au moins en théorie, des arguments diagnostiques en montrant :
-
- une silhouette cardiaque particulière, dite « en sabot », liée à la position surélevée de la pointe du cœur secondaire à l'hypertrophie ventriculaire droite ;
- un « coup de hache » à l'emplacement théorique du renflement lié à la présence du tronc pulmonaire, ici hypoplasique ;
- une vascularisation pulmonaire normale ou diminuée (« poumons clairs ») ;
- parfois, une position anormale du « bouton aortique » lié à la présence d'une crosse aortique à droite, particularité relativement fréquemment associée à une Tétralogie de Fallot (et qui influencerait le choix du coté où serait faite une anastomose palliative type Blalock-Taussig).
Il convient cependant de dire ici combien il est difficile de réaliser un bon cliché thoracique chez un nouveau-né. Lorsque les conditions requises (immobilité, inspiration forcée ...) ne sont pas réunies, ces signes peuvent être masqués ou pire totalement modifiés et induire en erreur. Un bon cliché thoracique n'est souvent obtenu que chez l'enfant plus grand ... alors que le diagnostic est établi depuis longtemps.
Autrefois indispensable, il n'est maintenant éventuellement pratiqué qu'avant la cure chirurgicale de la malformation pour s'assurer :
-
- de l'absence de communication interventriculaire (CIV) autre que la CIV sous-aortique constitutive de la malformation,
- de l'absence d'anomalie de naissance ou de trajet d'une artère coronaire (qui pourraient être lésée lors de l'intervention),
- de l'absence de rétrècissement(s) situés de façon plus distale sur les branches des artères pulmonaires,
- et faire le cas échéant, un bilan plus précis des autres anomalies éventuellement associées à la Tétralogie de Fallot.
Dans ses indications, le cathétérisme cardiaque est de plus en plus concurrencé par l'examen tomodensitométrique qui permet également mais plus simplement une étude fine de la circulation pulmonaire et des artères coronaires (coroscan).
Étiologie
Anomalie chromosomique
- micro délétion 22q11, à rechercher devant toute anomalie cono-troncale
- Syndrome de Down ou Trisomie 21
- Trisomie 13
- Trisomie 18
Tout en sachant que la malformation cardiaque le plus souvent observée dans ces trisomie est la présence d'une communication interventriculaire.
Syndromes
La tétralogie de Fallot est une malformation décrite dans plusieurs syndromes.
▼ Syndromes comportant une tétralogie de Fallot▼- Syndrome d'alcoolisme fœtal
- Syndrome de Goldenhar
- Syndrome TAR
- Syndrome de Yunis-Varon
- Acidurie méthacrylique
- Syndrome de Steinfeld
- Syndrome de Nager
- Syndrome de Sutherland Haan
- Syndrome de McKusick Kaufman
- Syndrome 3C
- Nanisme microcephalique osteodysplastique primordial type 1
- Acro-céphalo-syndactylie
- Syndrome d'Alagille
- Syndrome de Noonan
- Syndrome Kabuki
- Syndrome CHARGE
- Association VATER
- Distichiasis lymphœdème et fentes palatines
- Syndrome de Townes-Brocks
- Syndrome C
- Déficit en 17-bêta-hydroxystéroïde déshydrogénase
- Syndrome de Melnick-Needles
- Syndrome oto facio cervical
- Syndrome de Johnson Mcmillin
- Syndrome de Cayler
- Syndrome des yeux de chat
- Syndrome de Bindewald Ulmer Muller
- Microdélétion 22q11
Anomalies génétiques
En dehors des anomalies chromosomiques et des syndromes comportant cette pathologie, il exsite plusieurs gènes dont la mutation provoque une tétralogie de Fallot.
Géne MIM Chromosome Fréquence JAG1 601920 (en) 20p12 ? CSX 600584 (en) 5q34 ? ZFPM2 603693 (en) 8q23 ? Traitement
Traitement médical
Le traitement de la Tétralogie de Fallot, comme pour la plupart des malformations cardiaques, est un traitement chirurgical et non médical. Dans l'attente de la cure chirurgicale, on peut cependant être amené à proposer :
- Un apport de fer, plus ou moins systématique en cas de polyglobulie (excès de globules rouges), pour lutter contre la microcytose (petite taille des globules rouges);
- L'utilisation d'un traitement béta-bloquant par voie orale qui peut aider à prévenir les malaises anoxiques ou leur récidive (indication discutée). En cours de malaise, l'administration intra-veineuse d'un béta-bloquant facilite la levée du « spasme infundibulaire » à l'origine de l'accident (indication plus généralement admise).
Dans certaines formes qui n'ont pu faire l'objet d'un traitement chirurgical, on peut être conduit à proposer :
- Une oxygénothérapie continue ou discontinue, plusieurs heures par jour, par sonde nasale ou « lunettes »,
- La pratique de saignée en cas de polyglobulie importante et responsable de manifestations fonctionnelles (maux de tête, vertiges, fatigue importante ...). Il est habituellement admis qu'à elle seule, la présence d'une polyglobulie, quelle que soit son importance, ne justifie pas la réalisation de saignées.
Habituellement, la Tétralogie de Fallot n'entraine pas d'insuffisance cardiaque. Il n'y a donc aucune indication de traitement diurétique et/ou digitalique, ce dernier pouvant même aggraver la sténose infundibulaire et faciliter ainsi l'apparition de malaise. De même, l'administration de traitements vaso-dilatateurs (tels que les IEC ou Inhibiteur de l'enzyme de conversion de l'angiotensine) peut être non seulement inutile mais délétère en abaissant les résistances artérielles systémiques et en aggravant ainsi le shunt droit-gauche (passage de sang bleu - pauvre en oxygène du ventricule droit vers l'aorte).
Il est, comme dans toutes les cardiopathies congénitales, important de prévenir ou traiter dès le début toute infection afin d'éviter que se développe une infection à l'intérieur même du coeur (Endocardite infectieuse) ou un embole septique à point de départ veineux et à point d'arrivée dans un organe alimenté par l'aorte (rein, rate, foie et surtout cerveau). Historiquement, avant la chirurgie, les patients atteints de Tétralogie de Fallot mourraient le plus souvent soit au cours d'un malaise anoxique, soit par le développement d'un abcès cérébral.
Traitement chirurgical
Deux types d'intervention doivent être distingués: Les interventions palliatives qui visent à limiter les conséquences de la malformation mais ne la corrigent pas et l'intervention curatrice qui s'attache à rétablir une anatomie aussi normale que possible.
Chirurgie palliative
Elle n'est pas toujours nécessaire, la plupart des enfants présentant une Tétralogie de Fallot atteignant sans encombre l'âge de 4 à 12 mois auquel sera pratiquée l'intervention correctrice définitive. Une intervention palliative ne sera envisagée que si l'enfant présente une cyanose intense et précoce et/ou des malaises anoxiques.
Le plus souvent, l'intervention sera une anastomose faite entre l'aorte (ou une de ses branches) et l'artère pulmonaire pour améliorer l'apport sanguin dans les artères pulmonaires et donc l'oxygénation de tout l'organisme. Plusieurs types d'anastomose peuvent être envisagés et nous invitons le lecteur à se reporter pour plus de détails à l'article consacré à l' anastomose de Blalock-Taussig, la plus fréquemment pratiquée.
Nous citerons également ici la dilatation des valves pulmonaires, bien qu'il s'agisse d'un geste de cathétérisme interventionnel et non de chirurgie. Elle est parfois pratiquée chez le nourrisson quand la sténose valvulaire pulmonaire constitue l'obstacle prédominant (elle n'a pas d'effet sur la sténose infundibulaire, sinon qu'elle peut parfoir en ralentir la progression).
Chirurgie curatrice
Elle vise à fermer la communication interventriculaire et à rétablir le mieux possible la perméabilité de la voie pulmonaire. Ses modalités pourront être influencées par la présence de communications multiples, la sévérité de l'hypoplasie de la voie pulmonaire (fente de l'anneau pulmonaire, patch d'élargissement du tronc pulmonaire et de la bifurcation...) et la présence éventuelle d'une anomalie de trajet d'une artère coronaire.
Devenir des enfants
- Après anastomose palliative de type « Blalock-Taussig » (chirurgie palliative):
Les nourrissons ou petits enfants sont à l'abri des malaises anoxiques (mais non de la cyanose) et peuvent poursuivre un développement staturo-pondéral proche de la normale pendant plusieurs années. Bien faite, c'est-à-dire dérivant suffisamment mais pas trop de sang vers la circulation pulmonaire, cette seule anastomose peut permettre une survie prolongée (jusqu'à 50 ans dans une observation personnelle) mais elle ne protège pas d'autres complications, en particulier infectieuses et emboliques. Malgré ses limites, une anastomose de Blalock-Taussig (le plus souvent modifiée) reste une intervention très utile, en particulier dans les pays en voie de développement où la cure complète de la malformation (sous circulation extra-corporelle) ne peut être envisagée avant de nombreuses années ... quand elle peut l'être.
- Après « cure complète » (chirurgie curatrice):
Théoriquement, et en pratique de plus en plus souvent, il s'agit d'une intervention unique, définitive, obtenant une "guérison complète" et permettant une vie normale sans traitement médical, même si on considère actuellement que la Tétralogie de Fallot est une malformation non seulement cardiaque mais aussi de la vascularisation intra-pulmonaire et qu'à ce titre, elle restera responsable d'une certaine limitation à l'effort. Elle peut cependant comporter certaines complications à distance. La plus fréquente est l'aggravation de la fuite de la valve pulmonaire crée par le chirurgien et quasiment inéluctable. Celle ci peut nécessiter une réfection de la valve, soit par une nouvelle intervention, soit par mise en place d'une prothèse au cours d'un catéthérisme [3]. Des troubles du rythme cardiaque peuvent également se voir, avec rarement, des morts subites[6].
Conseil génétique
En l'absence d'anomalie chromosomique, le risque de récurrence est de 3.1 %[7] soit trois fois plus que le risque observé dans la population générale.
Sources
- (fr) Site en français de renseignement sur les maladies rares et les médicaments orphelins
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:187500 [1]
Références
- ↑ Stensen N, Embryo Monstro Affins Parisiis Dissectus, Acta Med Philos Hafniensa 1671–72;1:202–203
- ↑ Fallot E, Contribution a l’anatomie pathologique de la maladie bleu (cyanose cardiaque), Mars Med 1888;25(418)
- ↑ a et b Huehnergarth KV, Gurvitz M, Stout KK, Otto CM, Repaired tetralogy of Fallot in the adult: monitoring and management, Heart, 2008;94:1663-1669
- ↑ Pézard P, Bonnemains L, Boussion F, Sentilhes L, Allory P, Lépinard C, Guichet A, Triau S, Biquard F, Leblanc M, Bonneau D, Descamps P. Influence of ultrasonographers training on prenatal diagnosis of congenital heart diseases: a 12-year population-based study.Prenat Diagn. 2008 Nov;28(11):1016-22
- ↑ Taussig HB - Congenital malformations of the heart. Cambridge, the Commonwealth Fund, Harvard university Press, 1960, vols I and II
- ↑ Gatzoulis MA, Balaji S, Webber SA et als. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: a multicentre study, Lancet, 2000;356:975–81
- ↑ Burn J, Brennan P, Little J et als. Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study, Lancet, 1998;351):311–316
 Portail de la médecine
Portail de la médecine
Catégories : Médecine fœtale | Cardiopathie congénitale



Wikimedia Foundation. 2010.