- SEAr
-
Substitution électrophile aromatique
La substitution électrophile aromatique (ou SEA, voire SEAr) est une réaction chimique du domaine de la chimie organique, au cours de laquelle un atome, en règle générale d'hydrogène, fixé à un cycle aromatique est substitué par un groupement électrophile. C'est une réaction très importante en chimie organique, tant dans l'industrie qu'en laboratoire. Elle permet de préparer des composés aromatiques substitués par une grande variété de groupements fonctionnels suivant le bilan :
-
- ArH + EX → ArE + HX
avec Ar un composé aromatique et E un groupement électrophile.
Mécanisme général de la réaction
 Mécanisme général de la substitution électrophile aromatique
Mécanisme général de la substitution électrophile aromatique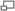
La première étape du mécanisme est une addition au cours de laquelle le composé électrophile A+ réagit avec un doublet électronique du cycle aromatique. Cette étape nécessite généralement une catalyse par un acide de Lewis. Cette addition conduit à la formation d'un carbocation cyclohexadiénil connu sous le nom d’intermédiaire de Wheland (ou complexe σ, ou encore ion arhénium). Ce carbocation est instable, puisqu'il correspond à la fois à la présence d'une charge sur la molécule et à une perte d'aromaticité. Il est néanmoins stabilisé par mésomérie : la charge est en réalité délocalisée sur plusieurs atomes de carbone.
Au cours de la seconde étape, la base conjuguée de l'acide de Lewis (ou un anion présent dans le milieu réactionnel, par exemple dans le cas de la sulfonation) attaque l'atome d'hydrogène lié au carbone ayant subi l'addition électrophile. L'électron qui était utilisé pour la liaison C-H permet alors au système de retrouver son aromaticité.
Principales substitutions électrophiles aromatiques
Ce chapitre détaille les principales substitutions électrophiles aromatiques utilisées dans l'industrie et en laboratoire. Pour chacune d'entre elles, le mécanisme de la réaction est donné dans le cas particulier du benzène. Ce mécanisme est similaire pour d'autres types de composés aromatiques, aux conditions opératoires (température, solvant...) près.
Nitration aromatique
La nitration aromatique est une substitution électrophile aromatique particulière au cours de laquelle un atome d'hydrogène lié à un atome de carbone du cycle aromatique est substitué par un groupement nitro -NO2 pour former du nitrobenzène. L'électrophile utilisé pour la substitution est NO2+ (ion nitronium), produit in-situ.
Dans la pratique pour effectuer la substitution, le benzène est chauffé à reflux (à 50 °C environ) dans un mélange d'acide sulfurique et d'acide nitrique. Le schéma réactionnel est le suivant :
-
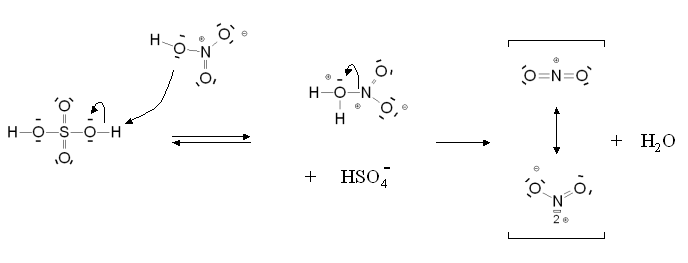
- (1) 2H2SO4 + HNO3 → 2HSO4- + NO2+ + H3O+
-
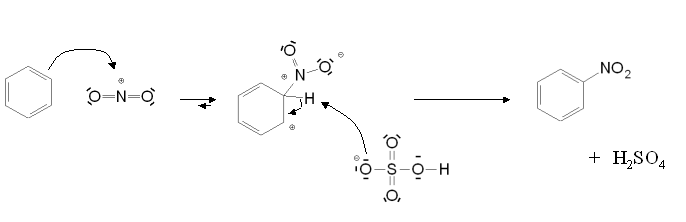
- (2) C6H6 + NO2+ → C6H5NO2 + H+
-
- (3) H+ + H3O+ + 2HSO4- → H3O+ + H2SO4 + HSO4-
L'acide sulfurique joue en quelque sorte le rôle de catalyseur pour la formation de l'ion nitronium. La réaction est également possible avec l'acide nitrique seul, mais elle est alors beaucoup plus lente. Parmi les autres réactifs utilisables pour la nitration aromatique, on peut citer le tétrafluoroborate de nitronium, qui est un sel de nitronium obtenu à partir de fluorure d'hydrogène, d'acide nitrique et de trifluorure de bore.
 Mécanisme de formation de l'ion nitronium
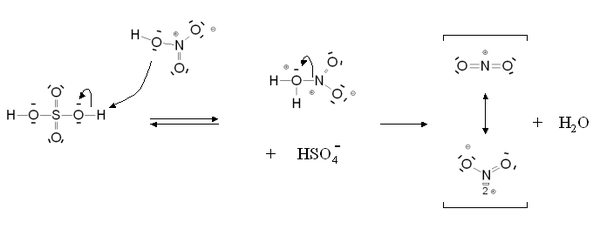
Mécanisme de formation de l'ion nitronium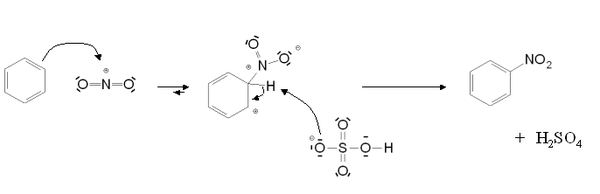 Mécanisme de la nitration aromatique
Mécanisme de la nitration aromatiqueSi la réaction est catalysée en présence d'acide sulfurique, l'étape cinétiquement déterminante est la nitration du cycle benzénique pour former l'intermédiaire de Wheland. En présence d'acide nitrique seul, il s'agit de la formation de l'ion nitronium.
Le nitrobenzène formé au cours de cette réaction peut notamment être utilisé pour fabriquer de l'aniline par réduction :
-
- C6H5NO2 + 3H2 → C6H5NH2
Sulfonation aromatique
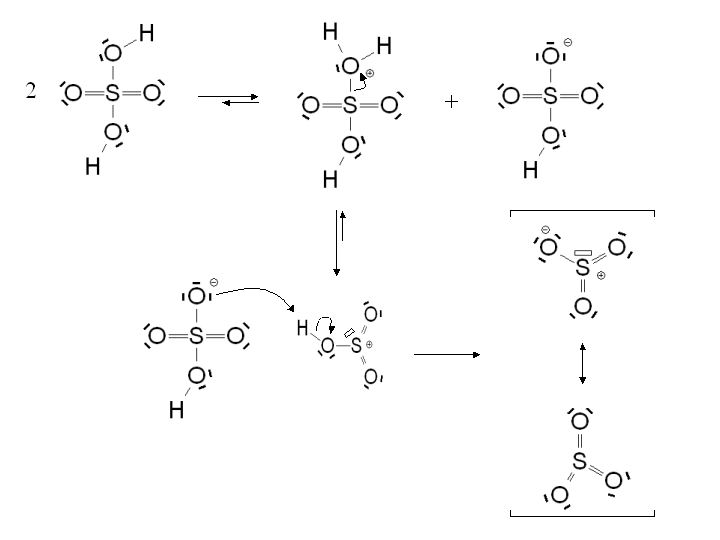
 Mécanisme de formation de SO3 dans l'acide sulfurique
Mécanisme de formation de SO3 dans l'acide sulfuriqueLa sulfonation aromatique est une substitution électrophile aromatique particulière au cours de laquelle un atome d'hydrogène lié à un atome de carbone du cycle aromatique est substitué par un groupement acide sulfonique. Dans le cas du benzène, la réaction permet de former de l'acide benzène sulfonique.
La substitution peut être réalisée de deux manières :
- Le benzène est maintenu à 25 °C dans un oléum, solution de SO3 dans l'acide sulfurique (H2SO4) ou mélange de SO3 et d'eau (avec SO3 majoritaire). Le bilan de la réaction est alors :
-
- C6H6 + SO3 → C6H5SO3H
- Le benzène est chauffé dans l'acide sulfurique concentré. SO3 est alors formé in-situ par réaction de l'acide sulfurique sur lui-même. Le bilan de la réaction est alors :
-
- C6H6 + 2 H2SO4 → C6H5SO3H
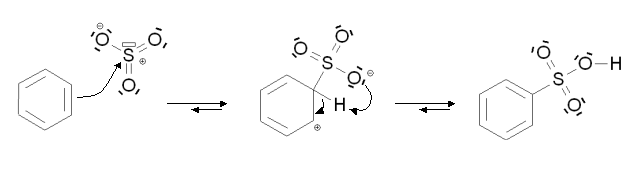
Dans les deux cas, le mécanisme de la réaction est le suivant :
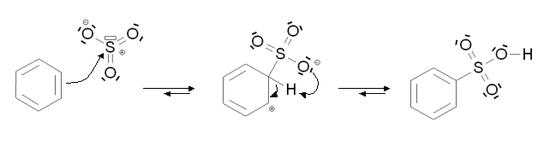
Pour la sulfonation, l'élimination de l'atome d'hydrogène se fait par une réaction intramoléculaire.
Cette réaction ne possède pas d'étape cinétiquement déterminante.
Il s'agit d'une réaction réversible : il est possible d'éliminer le groupement acide sulfonique et de régénérer le benzène en chauffant l'acide benzène sulfonique dans une solution diluée d'acide sulfurique dans de l'eau surchauffée. Le bilan est alors :
-
- C6H5SO3H + H2O(vapeur) → C6H6 + HSO4- + H3O+
L'acide benzène sulfonique formé au cours de cette réaction est un intermédiaire de synthèse important dans l'industrie, utilisé dans la fabrication de colorants et de produits pharmaceutiques. Par ailleurs, il est possible de le réduire en présence de soude fondue pour former du phénol.
Halogénation aromatique
L’halogénation aromatique est une substitution électrophile aromatique au cours de laquelle un atome d'hydrogène lié à un atome de carbone du cycle aromatique est substitué par un élément halogène suivant le bilan suivant :
-
- C6H6 + X2 → C6H5X + HX
La réaction n'est pas spontanée, mais nécessite la présence d'un catalyseur de type acide de Lewis. Elle s'effectue donc en milieu anhydre. Elle est possible sans catalyseur (mais est alors lente) dans le cas de cycles activés, comme par exemple le phénol. L'halogénation aromatique permet de substituer un atome d'hydrogène par un atome de chlore, de brome ou d'iode. En revanche, elle n'est pas possible avec le fluor. Celui-ci est en effet un oxydant puissant qui entraîne une dégradation du composé aromatique. Le mécanisme de la réaction est le suivant (exemple dans le cas d'une chloration) :
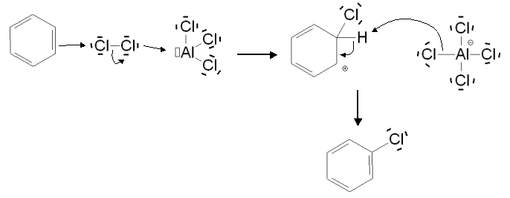
Au cours de la première étape du mécanisme, l'acide de Lewis utilisé comme catalyseur forme un complexe avec le dichlore, ce qui rend la liaison Cl-Cl polarisée. L'un des deux atomes de chlore devient donc électrophile, et peut subir l'attaque nucléophile du cycle aromatique, conduisant ainsi à la formation de l'intermédiaire de Wheland. L'anion formé contribue ensuite dans la deuxième étape à l'élimination de l'atome d'hydrogène et à la restauration de l'aromaticité.
Le catalyseur utilisé est généralement constitué du même élément halogène que celui agissant dans la substitution. Les acides de Lewis les plus couramment employées sont donc ZnCl2, AlCl3 et FeCl3 dans le cas de la chloration, et FeBr3 dans le cas de la bromation. Dans le cas de l'iode, le mécanisme de la réaction est légèrement différent. En effet, le diiode I2 est trop peu réactif. Il doit d'abord réagir avec un agent d'oxydation (par exemple l'acide nitrique) pour former l'ion I+, électrophile, qui interviendra dans l'iodation.
Les halogènes sont des éléments faiblement désactivant pour le cycle aromatique. En conséquence, si la réaction est catalysée et que l'halogène est présent en excès, il pourra se produire des polysubstitutions.
Réactions de Friedel-Crafts
Les réactions de Friedel-Crafts sont des substitutions électrophiles aromatiques particulières au cours desquelles un cycle aromatique est alkylé (substitution d'un atome d'hydrogène par un groupement alkyle) ou acylé (substitution d'un atome d'hydrogène par un groupement acyle).
- Article détaillé : Réaction de Friedel-Crafts.
Alkylation
L'alkylation de Friedel-Crafts est une réaction d'alkylation d'un composé aromatique. Cette réaction nécessite une catalyse par un acide de Lewis.
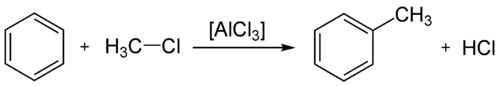 Bilan de l'alkylation de Friedel-Crafts
Bilan de l'alkylation de Friedel-CraftsAcylation
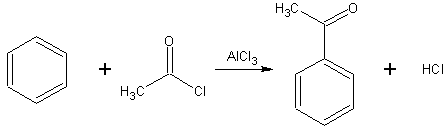
L'acylation de Friedel-Crafts est une réaction d'acylation d'un composé aromatique. Comme l'alkylation, elle nécessite une catalyse par un acide de Lewis. Les principaux catalyseurs utilisés sont le chlorure d'aluminium et le bromure d'aluminium. Il y a en général plus besoin de catalyseur que les quantités stoechiométriques, car il complexe avec le produit formé, d'où la nécéssité d'une hydrolyse après la réaction pour détruire le complexe.
 Bilan de l'acylation de Friedel-Crafts
Bilan de l'acylation de Friedel-CraftsAutres substitutions
Réaction de Kolbe-Schmitt
La réaction de Kolbe-Schmitt (ou procédé de Kolbe) est une réaction de carboxylation mise au point par A. Kolbe et R. Schmitt. Au cours de cette substitution électrophile aromatique, du phénolate de sodium (sel de phénol) est chauffé à 125 °C en présence de dioxyde de carbone sous une pression de 100 atm, puis traité par de l'acide sulfurique. Le bilan de la réaction est le suivant :
-
- C6H5OH + NaOH + H2SO4 → C6H4OHCOOH + HSO4-
 Mécanisme de la synthèse de Kolbe-Schmitt
Mécanisme de la synthèse de Kolbe-SchmittAu cours de la première étape (pas montrée sur le schéma), le phénol réagit avec de la soude pour former le phénolate de sodium et des ions OH-. Le phénolate réagit ensuite avec le dioxyde de carbone par substitution électrophile aromatique, le centre électrophile étant ici l'atome de carbone du CO2. Les ions OH- formés au cours de la première étape assistent la restauration de l'aromaticité. Le composé obtenu étant la base conjuguée de l'acide carboxylique, la dernière étape consiste en une réaction acide-base avec l'acide sulfurique.
Le produit obtenu au cours de cette réaction est un hydroxy-acide aromatique (ici l'acide salicylique, précurseur de l'aspirine).
Réactions avec des cycles aromatiques déjà substitués : les polysubstitutions
Le produit d'une réaction de substitution électrophile aromatique est lui-même un composé aromatique : l'élimination d'un atome d'hydrogène permet de restaurer l'aromaticité de l'intermédiaire de Wheland (le carbocation intermédiaire). Rien ne s'oppose donc à ce que ce produit, qui est un cycle aromatique substitué, subisse de nouveau une substitution électrophile aromatique, tant qu'il reste des atomes d'hydrogène liés à des atomes de carbone. En réalité, tous les composés aromatiques substitués ne pourront pas subir une nouvelle substitution électrophile aromatique, et le produit issu d'une seconde réaction sera dépendant du produit de départ : le groupement présent sur le composé substitué de départ influence à la fois la réactivité de ce composé (il peut ou non subir une seconde substitution), ainsi que la régiosélectivité de la réaction (tous les produits possibles ne sont pas formés).
Réactivité vis à vis de la polysubstitution
Le groupement présent sur le composé substitué de départ influence fortement sa réactivité. Ces groupements sont classés en deux catégories : des groupements activants et des groupements désactivants. Un composé aromatique substitué par un groupement activant est ainsi plus réactif que le composé aromatique non substitué. A l'inverse, un composé aromatique substitué par un groupement désactivant est moins réactif. Ces règles ont été énoncé par le chimiste Holleman en 1910, elles sont connues sous le nom de règles de Holleman.
Groupements activants
Un groupement activant est un groupement dont la présence augmente la réactivité du cycle aromatique vis à vis de la substitution électrophile aromatique par rapport au cycle pour lequel ce groupement est absent. Une substitution d'un cycle aromatique non substitué par un groupement activant conduira fréquemment à une polysubstitution. La réactivité du cycle aromatique sera augmentée s'il faut fournir moins d'énergie pour passer du composé de départ à l'intermédiaire réactionnel (intermédiaire de Wheland), autrement dit si l'écart énergétique entre le composé de départ et l'intermédiaire réactionnel est plus faible. Ce sera notamment le cas des composés mésomères donneurs, comme par exemple le groupement alcool -OH. La figure ci-dessous montre la stabilisation par mésomérie du phénol et de l'intermédiaire de Wheland correspondant :
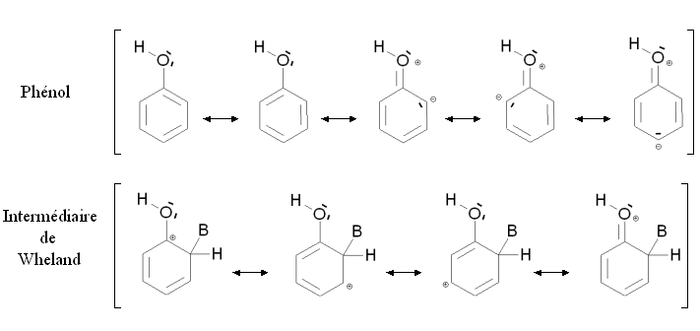
Le phénol et l'intermédiaire de Wheland sont tous deux stabilisés par mésomérie en délocalisant les électrons du cycle aromatique. Par ailleurs, il est possible d'écrire des formules mésomères mettant en jeu un doublet d'électron de l'atome d'oxygène. Dans le cas du phénol, ces formes mésomères font apparaître des charges formelles sur l'oxygène et un atome de carbone, elles n'induisent donc qu'une faible stabilisation. Au contraire, dans le cas de l'intermédiaire de Wheland, aucune charge supplémentaire n'apparaît : la stabilisation est importante. L'intermédiaire de Wheland est donc plus stabilisé que le phénol par la présence du groupement -OH.
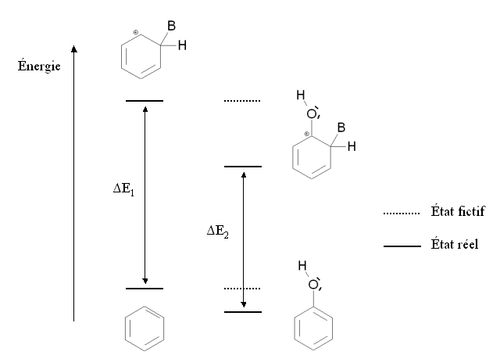 Écart énergétique entre le produit de départ et l'intermédiaire de Wheland pour le benzène et le phénol
Écart énergétique entre le produit de départ et l'intermédiaire de Wheland pour le benzène et le phénolCette situation est résumée sur la figure ci-contre, par comparaison avec la benzène (cycle aromatique non substitué). Les états fictifs (pointillés) correspondent à une situation où la présence du groupement -OH serait neutre, c’est-à-dire une situation où il n'interviendrait par l'intermédiaire des doublets portés par l'oxygène. Dans ce cas, l'énergie du phénol et de l'intermédiaire de Wheland serait la même que dans le cas du benzène. Les traits pleins correspondent à la situation réelle : l'intermédiaire de Wheland est plus stabilisé que le phénol (son énergie est plus abaissée). En conséquence, l'écart énergétique entre le phénol et l'intermédiaire de Wheland ΔE2 est plus faible que cet écart dans le cas du benzène ΔE1. Le phénol est donc plus réactif que le benzène vis à vis de la substitution électrophile aromatique.
Globalement, seront activants tous les groupements qui pourront stabiliser la charge positive de l'intermédiaire de Wheland, soit par mésomérie, soit par effet inductif, donc les groupements mésomères donneurs et inductifs donneurs.
Groupements désactivants
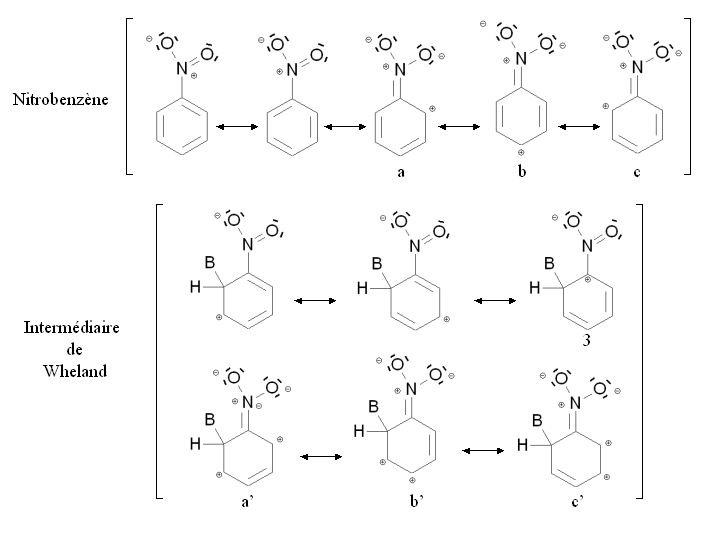
Un groupement désactivant est un groupement dont la présence diminue la réactivité du cycle aromatique vis à vis de la substitution électrophile aromatique par rapport au cycle pour lequel ce groupement est absent. Une substitution d'un cycle aromatique non substitué par un groupement désactivant ne conduira que très rarement à une polysubstitution. La réactivité du cycle aromatique sera diminuée s'il faut fournir plus d'énergie pour passer du composé de départ à l'intermédiaire réactionnel (intermédiaire de Wheland), autrement dit si l'écart énergétique entre le composé de départ et l'intermédiaire réactionnel est plus important. Ce sera notamment le cas des composés mésomères attracteurs, comme par exemple le groupement nitro -NO2. La figure ci-dessous montre la stabilisation par mésomérie du nitrobenzène et de l'intermédiaire de Wheland correspondant :
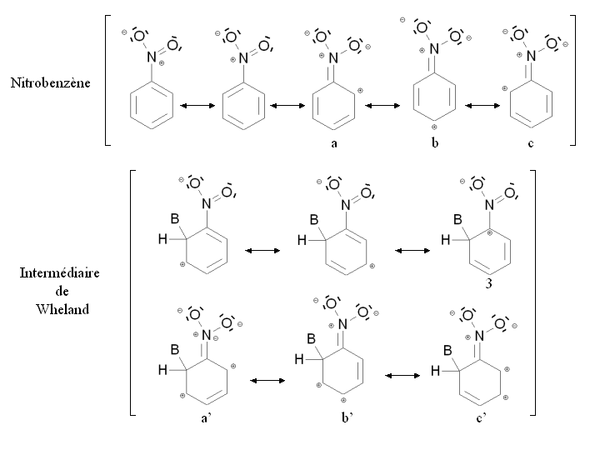
Le nitrobenzène et l'intermédiaire de Wheland sont tous deux stabilisés par mésomérie en délocalisant les électrons du cycle aromatique. Cependant dans le cas de la forme mésomère 3 de l'intermédiaire de Wheland, l'atome de carbone lié au groupement nitro (très électronégatif) est chargé positivement. Cette forme mésomère est donc très peu stabilisante : l'intermédiaire de Wheland est moins stabilisé que le nitrobenzène, et la présence du groupement nitro induit une déstabilisation par rapport à une situation où il serait absent. Par ailleurs, il est possible d'écrire des formules mésomères mettant en jeu un doublet d'électron de l'atome d'azote (a, b, c et a', b', c'). Cependant dans le cas des formes mésomères b' et c' de l'intermédiaire de Wheland, deux charges positives sont portées par des atomes de carbone conjoints, ce qui correspond à une configuration très peu stable. De nouveau, l'intermédiaire de Wheland est moins stabilisé que le nitrobenzène.
 Écart énergétique entre le produit de départ et l'intermédiaire de Wheland pour le benzène et le nitrobenzène
Écart énergétique entre le produit de départ et l'intermédiaire de Wheland pour le benzène et le nitrobenzèneCette situation est résumée sur la figure ci-contre, par comparaison avec la benzène (cycle aromatique non substitué). Les états fictifs (pointillés) correspondent à une situation où le groupement -NO2 n'interviendrait par l'intermédiaire des doublets portés par l'azote. L'état fictif de l'intermédiaire de Wheland est déstabilisé par rapport au benzène (à cause charge positive dans la formule mésomère 3). En revanche, le nitrobenzène n'est pas déstabilisé par rapport au benzène. Les traits pleins correspondent à la situation réelle : l'intermédiaire de Wheland est mois stabilisé que le nitrobenzène (son énergie est moins abaissée) à cause des charges portées par les atomes de carbone conjoints dans les formules mésomères b' et c'. En conséquence, l'écart énergétique entre le nitrobenzène et l'intermédiaire de Wheland ΔE2 est plus élevé que cet écart dans le cas du benzène ΔE1. Le nitrobenzène est donc moins réactif que le benzène vis à vis de la substitution électrophile aromatique.
Globalement, seront désactivants tous les groupements qui pourront déstabiliser la charge positive de l'intermédiaire de Wheland, soit par mésomérie, soit par effet inductif, donc les groupements mésomères attracteurs et inductifs attracteurs.
Bilan : réactivité relative de quelques composés substitués
La réactivité d'un composé aromatique substitué vis à vis d'une nouvelle substitution électrophile aromatique dépend donc fortement de la nature du substituant déjà présent. La réactivité est d'autant plus grande que le substituant apporte des électrons au système et stabilise les charges positives (effet mésomère donneur et inductif donneur). Le tableau ci-dessous donne ainsi quelques ordres de grandeurs de réactivité (rapportée à celle du benzène, fixée à 1) de quelques benzènes substitués. Le phénol est ainsi 1000 fois plus réactif que le benzène, et le nitrobenzène 10 000 fois moins.
substituant -N(CH3)2 -OH -CH3 -H -Cl -COOH -NO2 réactivité 9000000 1000 25 1 0,3 4.10-3 1.10-4 Régiosélectivité
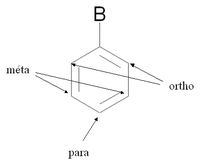 Nomenclature des positions du benzène substitué
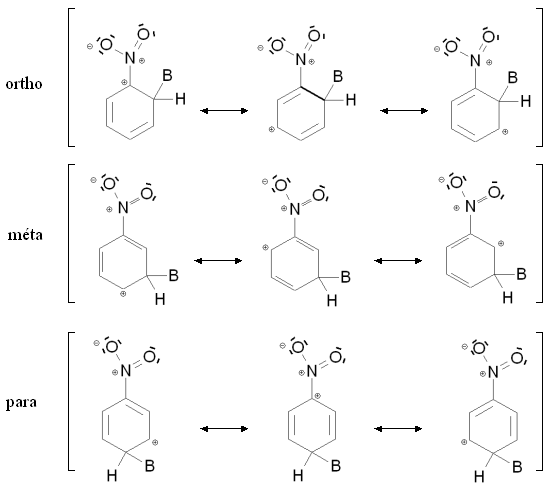
Nomenclature des positions du benzène substituéLorsqu'un composé aromatique substitué subit une seconde substitution électrophile aromatique, l'attaque peut a priori se faire depuis cinq positions. Parmi ces positions, deux sont des positions ortho, deux des positions méta et une position para (voir figure de droite).
De prime abord, on pourrait donc penser que le produit de la réaction est un mélange constitué à 40 % d'isomère ortho, à 40 % d'isomère méta et à 20 % d'isomère para, selon une répartition statistique (2 - 2 - 1). En réalité, ce n'est pas du tout le cas, et la régiosélectivité de la réaction (et donc la nature du produit final) dépend fortement du groupement déjà présent sur le cycle aromatique substitué. Suivant la nature de ce groupement, la seconde substitution pourra se faire quasi exclusivement en meta, ou selon un mélange ortho + para.
Groupements ortho-para orienteurs
Expérimentalement, une substitution électrophile aromatique utilisant comme produit de départ un cycle aromatique substitué par un groupement donneur (mésomère donneur ou inductif donneur) conduira à un mélange d'isomères ortho et para, avec une quantité très faible d'isomère méta. Ce résultat s'explique par des considérations énergétiques simples.
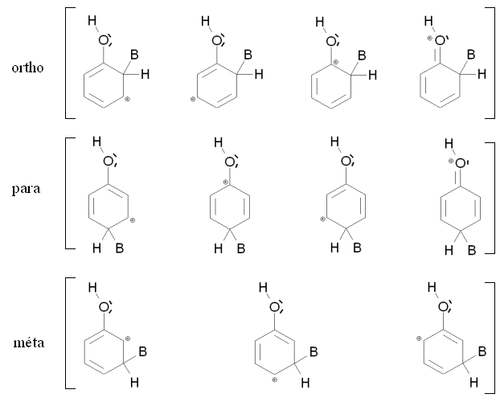 Stabilisation par mésomérie des intermédiaires de Wheland ortho, para et meta pour le phénol
Stabilisation par mésomérie des intermédiaires de Wheland ortho, para et meta pour le phénolPour passer du composé de départ à un intermédiaire réactionnel au cours d'une réaction chimique, il faut fournir de l'énergie pour franchir une barrière de potentiel. La vitesse de la réaction sera d'autant plus grande que cette barrière sera faible, et donc que l'intermédiaire réactionnel sera stable. Dans le cas de la substitution électrophile aromatique, on peut considérer avec une bonne approximation que le produit final formé le plus rapidement sera celui dont l'intermédiaire de Wheland sera formé le plus rapidement (réaction sous contrôle cinétique). Il doit donc correspondre à l'intermédiaire de Wheland le plus stable.
La figure de droite montre les intermédiaires de Wheland correspondant aux composés ortho, méta et para dans le cas où le produit de départ est le phénol (mésomère donneur). Dans les trois cas, l'intermédiaire de Wheland est stabilisé par mésomérie en délocalisant la charge positive sur trois atomes de carbone. Dans le cas des composés ortho et para, la charge est également stabilisée par une mésomérie mettant en jeu un doublet d'électrons de l'atome d'oxygène. Ces deux intermédiaires sont donc nettement plus stables que l'intermédiaire meta, et la réaction conduit à un mélange constitué principalement des isomères ortho et para.
Globalement, un groupement donneur (mésomère donneur ou inductif donneur) sera donc ortho para orienteur. Si ce groupement est très volumineux, le composé final sera majoritairement para (les positions ortho seront difficilement accessibles). Au contraire s'il est faiblement volumineux, la composé ortho sera statistiquement favorisé (2 positions ortho pour une seule position para). Ainsi par exemple, la nitration du toluène (φ-CH3) conduira à un mélange d'isomères ortho (60 %), para (37 %) et méta (2 %) (le substituant est peu volumineux), alors que la nitration du méthoxybenzène (φ-O-CH3) conduira à un mélange d'isomères ortho (34 %), para (65 %) et méta (1 %) (le substituant est relativement volumineux).
Groupements méta orienteurs
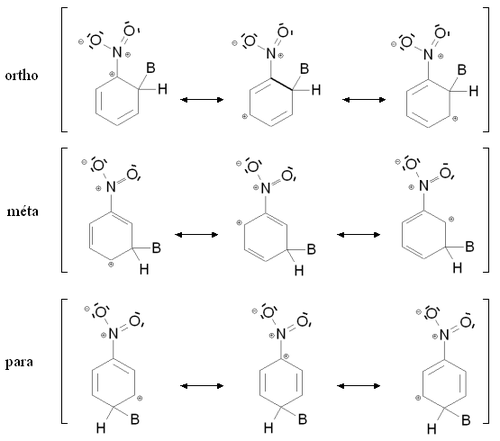 Stabilisation par mésomérie des intermédiaires de Wheland ortho, para et meta pour le nitrobenzène
Stabilisation par mésomérie des intermédiaires de Wheland ortho, para et meta pour le nitrobenzèneExpérimentalement, une substitution électrophile aromatique utilisant comme produit de départ un cycle aromatique substitué par un groupement attracteur (mésomère attracteur ou inductif attracteur) conduira principalement à l'isomère méta, avec une faible quantité d'isomères ortho et para.
Comme dans le cas des groupements ortho-para orienteurs, ce résultat s'explique par des considérations énergétiques simples en raisonnant sur la stabilité de l'intermédiaire de Wheland. La figure de droite montre les intermédiaires de Wheland correspondant aux composés ortho, méta et para dans le cas où le produit de départ est le nitrobenzène (mésomère attracteur). Dans les trois cas, l'intermédiaire de Wheland est stabilisé par mésomérie en délocalisant la charge positive sur trois atomes de carbone. Le groupement nitro étant attracteur, il ne stabilise jamais la charge positive par mésomérie. Pour l'une des formes mésomères des intermédiaires ortho et para, l'atome de carbone lié au groupement nitro porte une charge positive. Le groupement nitro étant très électronégatif, cette situation est très instable, et cette forme mésomère n'est quasiment pas stabilisante. Les intermédiaires ortho et para sont donc moins stables que l'intermédiaire méta, qui sera donc formé plus rapidement.
Globalement, un groupement attracteur (mésomère attracteur ou inductif attracteur) sera donc méta orienteur. Ainsi par exemple, une seconde nitration du nitrobenzène conduira à 92 % d'isomère méta, 7 % d'ortho et 1 % de para.
Cas particuliers des halogènes
Les halogènes (principalement chlore, brome et iode) constituent un cas particulier, étant à la fois faiblement mésomères donneurs et faiblement inductifs attracteurs. Ces groupements sont faiblement désactivants (le chlorobenzène est environ 3 fois moins réactif que le benzène) mais ortho-para orienteurs.
Tableau récapitulatif
En résumé, les groupements donneurs sont activants (la réactivité est plus importante) et ortho-para orienteurs, et les groupements attracteurs sont désactivants et méta orienteurs. En règle générale, l'effet activant ou désactivant est d'autant plus important que le groupement est plus donneur ou attracteur. Le tableau ci-dessous répertorie les effets sur la réactivité et la régiosélectivité de quelques groupements fréquemment utilisés.
Activant Désactivant Puissant -O-, -OH
ortho-para orienteurs-NO2, -NR3+ (R=H ou alkyle), -CCl3, -CF3
méta orienteursMoyens -O-R, -NH-CO-R (R=alkyle)
ortho-para orienteurs-CN, -SO3H, -COOR, -CO-R (R=H ou alkyle)
méta orienteursFaibles alkyles, aryles
ortho-para orienteurshalogènes
ortho-para orienteursSubstitutions mettant en jeu des composés hétérocycliques
Les composés aromatiques hétérocycliques, comme par exemple le furane, le pyrrole ou la pyridine, peuvent également réagir par substitution électrophile aromatique. Leurs comportements vis à vis de polysubstitutions (réactivité et régiosélectivité) sont déterminés par les mêmes considérations énergétiques que dans l'exemple du benzène.
Références
- Réaction de Kolbe-Schmitt
- (de) H. Kolbe, Annalen der Chemie und Pharmacie no 113, 1860, p. 125 ;
- (de) R. Schmitt, « Beitrag zu Kenntniss der Kolbe'schen Salicylsäure-Synthese », in Journal für praktische Chemie no 31 [2], 1885, p. 397 texte sur Gallica.
- Présentations de la SEA
- Peter Vollhardt, Traité de chimie organique, chap. 15-8 ;
- [pdf] La SEA, site de l'université Bordeaux I ; (lien périmé, à actualiser)
- [pdf] La SEA, site de l'université d'Orléans. (lien périmé, à actualiser)
Voir aussi
Articles connexes
 Portail de la chimie
Portail de la chimie

La version du 14 avril 2006 de cet article a été reconnue comme « article de qualité », c'est-à-dire qu'elle répond à des critères de qualité concernant le style, la clarté, la pertinence, la citation des sources et l'illustration. Catégories : Article de qualité | Réaction de substitution -


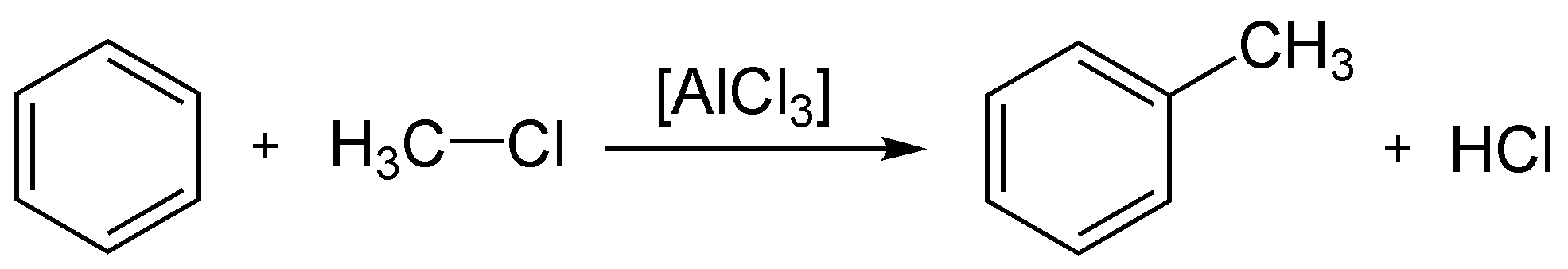






Wikimedia Foundation. 2010.