- Méthode de cluster couplé
-
Méthode du cluster couplé
Méthodes numériques pour le calcul de la structure électronique
Hartree-Fock Théorie de la perturbation de Møller-Plesset Interaction de configuration Méthode du cluster couplé Champ multi-configurationnel auto-cohérent Théorie de la fonctionnelle de la densité La méthode du cluster couplé, ou théorie du cluster couplé (expression souvent abrégée en « cluster couplé », en anglais coupled cluster) est une technique numérique de description des systèmes à plusieurs corps. Son utilisation la plus répandue est comme méthode ab initio de chimie quantique post-Hartree-Fock en chimie numérique. Il est basé sur la méthode d'orbitale moléculaire Hartree-Fock et lui ajoute un terme de correction afin de prendre en compte la corrélation électronique. Certains des calculs les plus précis pour des molécules de petite ou de taille moyenne utilisent cette méthode.
La méthode fut développée initialement par Fritz Coester et Hermann Kümmel dans les années 1950 afin d'étudier les phénomènes de physique nucléaire, mais devint plus fréquemment utilisée après que Jiři Čížek et Josef Paldus eurent reformulé la méthode pour l'adapter à la corrélation électronique dans les atomes et molécules dans les années 1960. Elle constitue à ce jour une des méthodes les plus répandues en chimie quantique incluant la corrélation électronique.Sommaire
Ansatz de fonction d'onde
La théorie de cluster couplé donne une solution approchée à l'équation de Schrödinger dépendante du temps
où
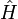 est le hamiltonien du système. La fonction d'onde et l'énergie de l'état de plus basse énergie sont notés respectivement
est le hamiltonien du système. La fonction d'onde et l'énergie de l'état de plus basse énergie sont notés respectivement 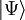 et E. D'autres versions de la théorie de cluster couplé, comme la méthode de cluster couplé de l'équation de mouvement ou la méthode de cluster couplé multi-références peuvent aussi donner des solutions approchées des états excités du système.
et E. D'autres versions de la théorie de cluster couplé, comme la méthode de cluster couplé de l'équation de mouvement ou la méthode de cluster couplé multi-références peuvent aussi donner des solutions approchées des états excités du système.
La fonction d'onde de la théorie du cluster couplé est écrite comme un ansatz exponentiel :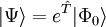 ,
,
où
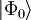 est un déterminant de Slater construit habituellement à partir des orbitales moléculaires Hartree-Fock.
est un déterminant de Slater construit habituellement à partir des orbitales moléculaires Hartree-Fock. 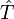 est un opérateur d'excitation qui, lorsqu'il agit sur , produit une combinaison linéaire des déterminants de Slater (voir ci-dessous pour de plus amples détails).
est un opérateur d'excitation qui, lorsqu'il agit sur , produit une combinaison linéaire des déterminants de Slater (voir ci-dessous pour de plus amples détails).
Le choix de l'ansatz exponentiel est opportun car (contrairement à d'autres, par exemple, l'interaction de configuration) il garantit l'extensivité de taille de la solution. La consistance de taille, dans la méthode de cluster couplé, dépend de la consistance de taille de la fonction d'onde référence.L'opérateur cluster
L'opérateur cluster est écrit sous la forme :
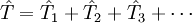 ,
,
où
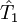 est l'opérateur de toutes les excitations singulières,
est l'opérateur de toutes les excitations singulières, 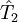 est l'opérateur de toutes les excitations doubles et ainsi de suite. Dans la formalisme de la seconde quantification ces opérateurs d'excitation sont exprimés de manière adéquate comme :
est l'opérateur de toutes les excitations doubles et ainsi de suite. Dans la formalisme de la seconde quantification ces opérateurs d'excitation sont exprimés de manière adéquate comme :et ainsi de suite.
Dans la formule ci-dessus,
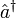 et
et 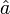 indiquent les opérateurs de création et de disparition respectivement, i, j sont utilisés pour les orbitales occupées et a, b pour les inoccupées. Les opérateurs de création et de disparition dans les termes de clusters couplés ci-dessous sont écrits dans une forme canonique, dans laquelle chaque terme est en ordre normal. Comme opérateurs d'excitation à une particule et à deux particules, et transforment la fonction de référence en une combinaison linéaire des déterminants de Slater simplement et doublement excités, respectivement. La résolution des coefficients inconnus
indiquent les opérateurs de création et de disparition respectivement, i, j sont utilisés pour les orbitales occupées et a, b pour les inoccupées. Les opérateurs de création et de disparition dans les termes de clusters couplés ci-dessous sont écrits dans une forme canonique, dans laquelle chaque terme est en ordre normal. Comme opérateurs d'excitation à une particule et à deux particules, et transforment la fonction de référence en une combinaison linéaire des déterminants de Slater simplement et doublement excités, respectivement. La résolution des coefficients inconnus 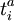 et
et 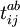 est nécessaire pour trouver la solution approchée .
est nécessaire pour trouver la solution approchée .
Si l'on prend en compte la structure de, l'opérateur exponentiel 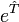 peut être développé en série de Taylor :
peut être développé en série de Taylor :Cette série est finie en pratique car le nombre d'orbitales moléculaires occupées est fini, comme d'ailleurs le nombre d'excitations. Afin de simplifier la recherche des coefficients t, le développement de
en opérateurs d'excitation individuels est achevé au deuxième ou à un niveau peu élevé d'excitation (rarement au delà de quatre). Cette approche est garantie par le fait que même si le système admet plus de quatre excitations, la contribution de 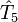 ,
, 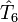 , etc. à l'opérateur est faible. De plus, si le plus haut niveau d'excitation dans les opérateurs est n,
, etc. à l'opérateur est faible. De plus, si le plus haut niveau d'excitation dans les opérateurs est n,alors les déterminants de Slater excités plus de n fois peuvent (et habituellement le font) contribuent encore à la fonction d'onde
en raison de la nature non-linéaire de l'ansatz exponentiel. Ainsi, le cluster couplé terminé à 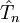 recouvre habituellement plus d'énergie de corrélation que l'interaction de configuration avec n excitations maximales.
recouvre habituellement plus d'énergie de corrélation que l'interaction de configuration avec n excitations maximales.Équations de cluster couplé
Les équations de cluster couplé sont des équations dont la solution est un ensemble de coefficients t. Il existe plusieurs façons d'écrire de telles équations mais le formalisme standard conduit à un ensemble final d'équations pouvant être résolu de manière itérative. L'approche variationnelle simple ne tire pas avantage de la nature connectée des amplitudes de clusters et conduit à un ensemble d'équations non achevé.
Supposons qu'il y ait q coefficients t afin de procéder à cette résolution : on a alors besoin de q équations. Il est aisé de noter que chaque coefficient t peut être mis en correspondance avec un certain détermant d'excitation :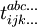 , qui correspond au déterminant obtenu à partir de par substitution des orbitales occupées i,j,k,... par les orbitales virtuelles a,b,c,.... En projetant l'équation de Schrödinger avec l'ansatz exponentiel par q différents déterminants par la gauche, on obtient les q équations recherchées :
, qui correspond au déterminant obtenu à partir de par substitution des orbitales occupées i,j,k,... par les orbitales virtuelles a,b,c,.... En projetant l'équation de Schrödinger avec l'ansatz exponentiel par q différents déterminants par la gauche, on obtient les q équations recherchées :où par
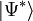 on entend l'ensemble complet des déterminants excités appropriés.
on entend l'ensemble complet des déterminants excités appropriés.Malheureusement,
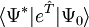 est une série inachevée. Les équations de cluster couplé sont réduites à une forme proche dans la représentation de similarité transformée :
est une série inachevée. Les équations de cluster couplé sont réduites à une forme proche dans la représentation de similarité transformée :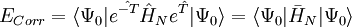 ,
,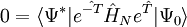 ,
,
les dernières étant les équations à résoudre et les précédentes l'équation d'évaluation de l'énergie. Considérant la méthode standard CCSD :
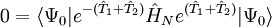 ,
,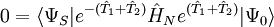 ,
,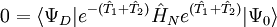 ,
,
qui lorsqu'elle est factorisé en utilisant la formule de Baker-Campbell-Hausdorff pour le hamiltonien transformé de similarité
![e^{\hat{-T}} \hat{H} e^{\hat{T}} = H + [H,T] + (1/2)[[H,T],T] + ...](/pictures/frwiki/102/ffa0ca40683364a985aad7bb96b1b3f1.png) , devient de degré 4 pour T1 et quadratique pour T2. Des codes standard de chimie quantique comme ACES ou NWChem résolvent les équations couplées en utilisant Ax = 0, où A est le jacobien du cluster couplé et x est le vecteur des amplitudes. En général, le hamiltonien transformé de similarité peut ne pas être hermitien.
, devient de degré 4 pour T1 et quadratique pour T2. Des codes standard de chimie quantique comme ACES ou NWChem résolvent les équations couplées en utilisant Ax = 0, où A est le jacobien du cluster couplé et x est le vecteur des amplitudes. En général, le hamiltonien transformé de similarité peut ne pas être hermitien.Types de méthodes de clusters couplés
La classification des méthodes de clusters couplés repose sur le nombre le plus élevé d'excitations permises dans la définition de
. Les abréviations utilisées pour les désigner commencent habituellement avec les lettres CC (pour cluster couplé - en anglais coupled cluster) suivies par :- S - pour les excitations simples - en anglais single excitation, raccourcies en singles dans la terminologie des clusters couplés.
- D - pour les excitations doubles - en anglais double excitation, raccourcies en double dans la terminologie des clusters couplés.
- T - pour les excitations triples - en anglais triple excitation, raccourcies en triple dans la terminologie des clusters couplés.
- Q - pour les excitations quadruples - en anglais quadruple excitation, raccourcies en quadruple dans la terminologie des clusters couplés.
Par conséquent, l'opérateur
en CCSDT est de la forme :Les termes entre parenthèses indiquent qu'ils sont calculés en se basant sur la théorie de la perturbation. Ainsi, une approche CCSD(T) signifie simplement :
- Méthode de cluster couplé,
- incluant des simples et double perturbations totalement prises en compte,
- les triples perturbations étant calculées par la théorie de la perturbation.
Description générale de la théorie
La complexité des équations et des codes de calcul correspondants, ainsi que le coût de calcul augmentent rapidement avec le niveau le plus élevé d'excitation. Pour de nombreuses applications, une précision suffisante peut être obtenue par CCSD, et la CCSD(T) plus précise (et plus coûteuse) est parfois appelée « l'étalon-or de la chimie quantique » pour son excellent compromis entre la précision et le coût pour des molécules proches des géométries d'équilibre. Des méthodes plus complexes comme la CCSDT et la CCSDTQ sont utilisées seulement pour des calculs de haute précision sur de petites molécules. L'inclusion de tous les n niveaux d'excitation pour un système à n électrons donne la solution exacte de l'équation de Schrödinger dans la base donnée.
Une amélioration possible pour une approche de cluster couplé standard est l'addition de termes linéaires dans les distances inter-électroniques par des méthodes comme CCSD-R12. Cela améliore le traitement de la corrélation électronique dynamique en satisfaisant la condition de col de Kato et accélère la convergence avec la base d'orbitales. Malheureusement, les méthodes R12 nécessitent la résolution de l'identité qui nécessite une base relativement importante afin d'être valides.
La méthode de cluster couplé décrite ci-dessus est connue comme la méthode de cluster couplé mono-référence (en anglais single-reference (SR) coupled-cluster method car l'ansatz exponentiel n'utilise qu'une fonction de référence. Les généralisations standard de la méthode SR-CC sont les approches multi-références (MR) : la méthode de cluster couplé d'état universel (state-universal coupled cluster en anglais, connu aussi comme la méthode cluster couplé dans un espace de Hilbert - Hilbert space coupled cluster), la méthode de cluster couplé de valence universelle (en anglais valence-universal coupled cluster, ou méthode de cluster couplé dans un espace de Fock - Fock space coupled cluster) et la méthode de cluster couplé sélective d'état (en anglais state-selective coupled cluster, ou encore méthode de cluster couplé d'état spécifique - state-specific coupled cluster).Une référence historique
Dans la première référence ci-dessous, H.G. Kümmel indique :
« Considering the fact that the CC method was well understood around the late fifties it looks strange that nothing happened with it until 1966, as Jiři Čížek published his first paper on a quantum chemistry problem. He had looked into the 1957 and 1960 papers published in Nuclear Physics by Fritz and myself. I always found it quite remarkable that a quantum chemist would open an issue of a nuclear physics journal. I myself at the time had almost gave up the CC method as not tractable and, of course, I never looked into the quantum chemistry journals. The result was that I learnt about Jiři's work as late as in the early seventies, when he sent me a big parcel with reprints of the many papers he and Joe Paldus had written until then. »-
« Considérant que la méthode CC était bien comprise à la fin des années cinquante environ, il semble étrange qu'il ne se soit rien produit avec elle jusqu'en 1966, quand Jiři Čížek a publié sont premier papier sur un problème de chimie quantique. Il avait cherché dans les articles de 1957 et 1960 publiés dans Nuclear Physics par Fritz et moi-même. J'ai toujours trouvé qu'il était assez remarquable qu'un chimiste quanticien pourrait ouvrir une voie à partir d'un journal de physique nucléaire. J'avais moi-même quasiment abandonné la méthode CC à cette époque, comme non tractable, et, bien sûr, je n'ai jamais cherché dans les journaux de chimie quantique. Le résultat fut que je n'ai pris connaissance du travail de Jiři qu'au début des années soixante-dix, lorsqu'il m'envoya un gros colis avec les copies des nombreux papiers que lui et Joe Paldus avaient écrit jusqu'alors. »
Théories liées
- Interaction de configuration
- Approximation de paire d'électrons couplés
Références
- H.G. Kümmel, A biography of the coupled cluster method - found in R.F. Bishop, T. Brandes, K.A. Gernoth, N.R. Walet, Y. Xian (Eds.), Recent progress in many-body theories, Proceedings of the 11th international conference, World Scientific Publishing, Singapore, 2002, pp. 334-348.
- Christopher J. Cramer, Essentials of Computational Chemistry, John Wiley & Sons, Ltd., 191 - 232 p. (ISBN 0-471-48552-7)
Ressources externes
- (en) Revue théorique et introduction à la théorie du cluster couplé
- Cramer, C. J. Essentials of Computational Chemistry: Theories and Models (Wiley and Sons)
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « Coupled cluster ».
 Portail de la chimie
Portail de la chimie Portail de la physique
Portail de la physique
Catégories : Chimie numérique | Chimie quantique
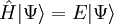
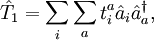
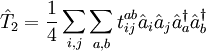
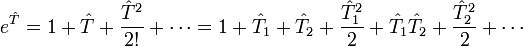
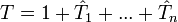
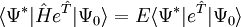
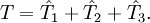
Wikimedia Foundation. 2010.