- Méthode de hartree-fock
-
Méthode de Hartree-Fock
Méthodes numériques pour le calcul de la structure électronique
Hartree-Fock Théorie de la perturbation de Møller-Plesset Interaction de configuration Méthode du cluster couplé Champ multi-configurationnel auto-cohérent Théorie de la fonctionnelle de la densité La méthode de Hartree-Fock est une méthode de résolution approchée de l'équation de Schrödinger d'un système quantique à N fermions utilisant le principe variationnel dans laquelle la fonction d'onde approchée est écrite sous la forme d'un déterminant de Slater :
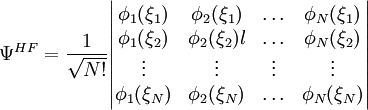
Les spinorbitales Φi(ξi) sont les solutions d'un système d'équations différentielles couplées appelées équations de Hartree-Fock :
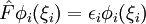
où
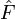 est l'opérateur de Fock. Dans le cas des atomes et des molécules, l'opérateur de Fock a pour expression :
est l'opérateur de Fock. Dans le cas des atomes et des molécules, l'opérateur de Fock a pour expression :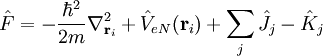
L'opérateur
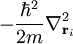 correspond à l'énergie cinétique de l'électron i. L'opérateur
correspond à l'énergie cinétique de l'électron i. L'opérateur 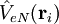 décrit le potentiel électrostatique entre cet électron et le(s) noyau(x). L'opérateur
décrit le potentiel électrostatique entre cet électron et le(s) noyau(x). L'opérateur 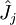 ou opérateur coulombien représente le potentiel moyen créé par les autres électrons et
ou opérateur coulombien représente le potentiel moyen créé par les autres électrons et 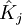 , l'opérateur d'échange, la correction à ce potentiel due à l'antisymétrie.
, l'opérateur d'échange, la correction à ce potentiel due à l'antisymétrie.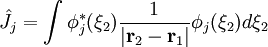
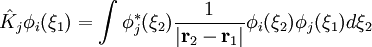
La méthode de Hartree-Fock est une approximation de champ moyen à particules indépendantes. L'opérateur de Fock dépend explicitement de ses solutions. La méthode de résolution la plus utilisée est la méthode du champ auto cohérent. Il s'agit d'une méthode itérative où l'opérateur de Fock est mis à jour à chaque itération avec les spinorbitales calculées à l'itération précédente. La calcul est arrêté lorsqu'une convergence satisfaisante (sur l'énergie, la fonction d'onde, .. etc.) est obtenue.
Le théorème de Koopmans donne aux valeurs propres εi de l'opérateur de Fock le sens physique d'opposé du potentiel d'ionisation
Pi = − εi Les fonctions d'onde Hartree-Fock satisfont le théorème d'Hellmann-Feynman et le théorème du viriel.
Voir aussi les applications de ce type de méthodes en physique nucléaire.
 Portail de la physique
Portail de la physique Portail de la chimie
Portail de la chimie
Catégories : Chimie quantique | Mécanique quantique | Physique quantique
Wikimedia Foundation. 2010.