- Thymine DNA glycosylase
-
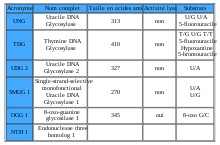 Figure 1: Exemple de membres de la famille des glycosylases humaines.
Figure 1: Exemple de membres de la famille des glycosylases humaines.
La thymine DNA glycosylase ou G/T mismatch-specific thymine DNA glycosylse (TDG) est une enzyme. Chez l'homme c'est une protéine monomérique de 410 acides aminés codés par 1233 paires de bases sur le chromosome 12 locus 24.1[1]. C’est une protéine qui agit à plusieurs niveaux de régulation mais qui n’a qu’une fonction catalytique : la réparation de l’ADN.
Sommaire
Utilisation
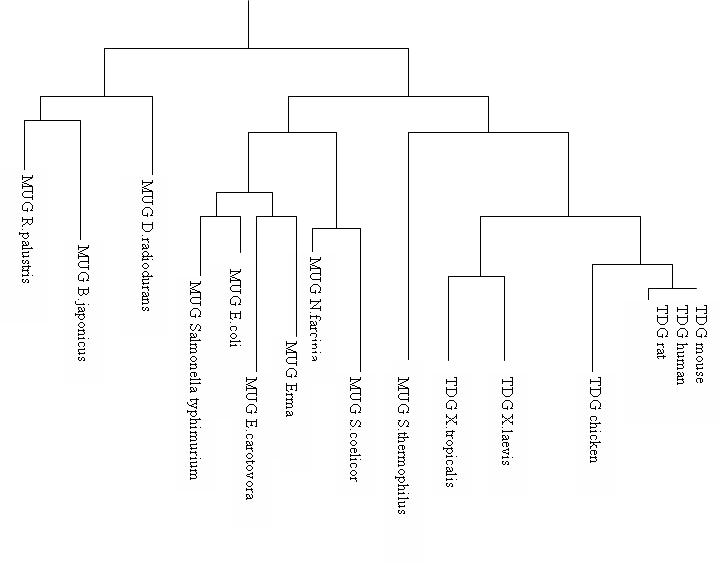
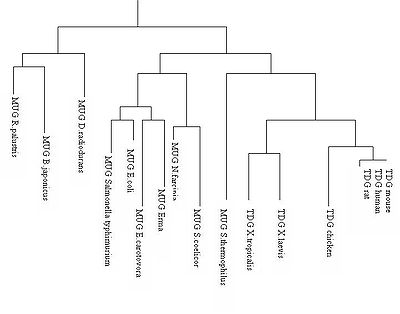 Arbre phylogénétique des glycosylases
Arbre phylogénétique des glycosylasesDepuis les découvertes des glycosylases d'ADN dans les années 1975, les variations de la thymine DNA glycosylase est utilisée pour déterminer la parenté dans l'arbre phylogénétique des vertébrés car une certaine homologie est gardée à travers le temps et les différentes espèces. Ceci est caractérisé par la conservation du site catalytique de la TDG. L'uracile DNA glycosylase (MUG) joue le même rôle dans les autres embranchements. Il semblerait par exemple que la MUG Escherichia coli possède 37% d’homologie avec la TDG humaine[2].
Les substrats de la TDG
La thymine DNA glycosylase ne peut enlever un couple de bases A/T par exemple, puisqu’il n’est pas un mésappariement[3]. De plus, la TDG est incapable de réparer un simple brin d’ADN, seulement l’ADN double brin. Elle reconnaît et signale préférentiellement les thymines et uraciles des mésappariements T/G et U/G, souvent créés par des désaminations spontanées de la cytosine ou de la 5-méthylcytosine[4]. La TDG est aussi en mesure d’exciser l’uracile et le 5-bromouracile. De plus, certains traitements contre le cancer sont inefficaces étant donné l’habileté de la TDG à substituer le 5-fluorouracile. Il semblerait que cette glycosylase ait la capacité de réparer les bases endommagées liées avec des thymines et des cytosines, mais cette hypothèse est plutôt controversé[5].
Les principes de réparation de l'ADN
Article détaillé : Réparation de l'ADN.Au cours de sa vie, une cellule, peu importe sa provenance, est confrontée à des événements susceptibles de créer des erreurs de réplication ou encore des mutations dans son ADN. Ces erreurs sont possiblement créées par la désamination, l’oxydation ou l’alkylation des bases azotées. De plus, les rayons X ou les UV ont la capacité d’endommager l’ADN. De nombreuses méthodes sont utilisées par la cellule pour réparer son ADN et celle utilisée, ainsi que les enzymes impliqués dépendent entre autres de l’importance de la mutation. En effet, ces dernières ne créent pas un même degré de distorsion de la double hélice lorsqu’elles se produisent. Un simple mésappariement d’une base azotée ne produit pas une distorsion de l’ADN suffisante pour être reconnue par tous les enzymes de réparation. Par contre, la thymine DNA glycosylase est en mesure de détecter cette erreur et de la réparer.
On dit de la TDG qu’elle est une glycosylase monofonctionnelle c’est-à-dire qu’elle hydrolyse le lien N-glycosydique entre le squelette de désoxyribose phosphate et la base endommagée afin de créer un site abasique ou « site AP ». Elle est incapable de couper le squelette lui-même, d’autres enzymes auront cette tâche.
Afin de contrer certains mésappariements de l’ADN, la TDG utilise la voie du « Base excision repair » (BER), contrairement à d’autres enzymes de réparation qui peuvent utiliser la voie du « Nucleotid excision repair » (NER). La TDG est en mesure d’exciser une base azotée endommagée en suivant le principe du « short patch repair » une des voies du BER.
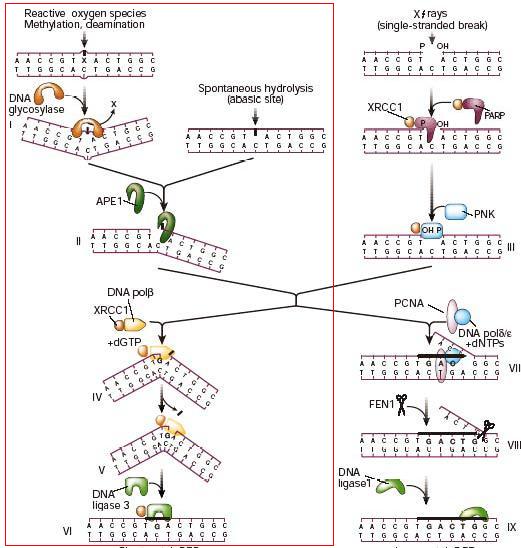
 Réparation de l'ADN par le principe d'excision d'une base endommagée[6], "short patch repair".
Réparation de l'ADN par le principe d'excision d'une base endommagée[6], "short patch repair".Premièrement, lorsqu’il y a une base azotée endommagée dans un brin double d’ADN, la Thymine DNA glycosylase la reconnaît et signale l’emplacement de la mutation. La TDG hydrolyse le lien N-glycosidique par lequel la base est attachée au squelette de sucre et la mutation est enlevée, le site AP est créé. Lorsque la TDG dégage la base endommagée, une forte affinité s’installe entre l’enzyme et la guanine restante en face du site AP. L’endonucléase APE-1, en s’approchant de la TDG, permettra de diminuer cette affinité afin de déloger la TDG du site AP. L’APE-1 pourra ensuite effectuer le clivage du nucléotide abasique. La Pol β viendra ensuite polymériser le nouveau nucléotide et la ligase 3 terminera la réparation. Xeroderma pigmentosum
Article détaillé : Xeroderma pigmentosum.La Xeroderma pigmentosum (XP) est maladie autosomique récessive rare, qui se caractérise par une sensibilité accrue aux rayons U.V. du soleil chez les patients homozygotes pour la mutation et même chez les hétérozygotes. Certaines personnes seront atteintes par une dégénérescence du système nerveux, environ 20%, et beaucoup seront atteints par les troubles ophtalmologiques. Quoi qu’il en soit, la grande majorité seront touchés par des carcinomes de la peau et des mélanomes[7]. Une personne sur 500 aurait un allèle du gène muté.
La protéine en cause dans cette affectation est la XPC, une enzyme de réparation de l’ADN impliquée dans le NER. Des études[8] tendent à prouver que la XPC, complexé avec la HR23B, joue aussi un rôle dans la voie du BER. En effet, le complexe XPC-HR23B tiendrait un rôle semblable à la APE-1 afin de libérer la TDG du site AP, site pour lequel elle a trop d’affinités.
Les mutations observées[9] sur le gène XPC sont créées par des non-sens, des décalages du cadre de lecture ou encore des anomalies dans le processus d’épissage. De plus, il semble qu’environ 8 gènes soient impliqués dans la Xeroderma pigmentosum. Par exemple, des mutations sur des oncogènes tel que RAS et des gènes suppresseurs de tumeurs tel que p53 ont été découvertes dans des fibroblastes XP[7].
Il parait évident que si la XCP-HR23B n’est plus en mesure d’exercer son influence sur la TDG, cette dernière s’accrochera à la guanine du site AP plus longtemps, altérant son efficacité de réparation. Les mutations de l’ADN n’étant plus excisées, la cellule devra utiliser les voies de signalisation bloquant le cycle cellulaire afin de ne pas se diviser en présence d’ADN erroné. Par contre, si ces voies, incluant p53, sont elles aussi touchées par des mutations dans leur propre ADN, la cellule ne possède plus les mécanismes nécessaires pour se diviser normalement et elle risque de devenir une cellule transformée, formant un cancer.
Voir aussi
Articles connexes
Références
- [réf. souhaitée]http://www.pubmed.com
- Hardeland, U., Bentele, M., Jiricny, J., Schär, P., 2003. The versatile thymine DNA glycosylase: a comparative characterisation of the human, Drosophila and fission yeast orthologs. Nucleic Acids Research, Vol.31,No.9, p.2261-2271.
- Yoon, J-H., Iwai, S., O’Connor, T.R., Pfeifer, G.P., 2003. Human thymine DNA glycosylase and methyl-CpG-binding protein 4 (MBD4) excise thymine glycol (Tg) from a Tg:G mispair. Nucleic Acids Research, Vol.31, No.18, p.5399-5404.
- Abu, M., Waters, T.R., 2003, The main role of human Thymine DNA glycosylase is removal of thymine produced by desaminaion of 5-methylcytosine and not removal of ethenocytosine. The journal of biological chemistry. Vol.278, No.10, p.8739-8744.
- Mohan,R.D., Rao,A., Gagliardi,J. and Tini,M., 2007,SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol.Cell. Biol. Vol.27, p.229-243.
- http://idg.u-strasbg.fr/PDFcours/cours_VS_2006.pdf
- point_scientifique
- Shimizu, Y., Iwai, S., Hanaoka, F., Sugasawa, K. 2003, Xeroderma pigmentosum group C protein interact physically and functionally eith thymine DNA glycosylase. The EMBO journal, Vol.22, No.1, p.164-173.
- Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients - Khan et al. 27 (1): 84 - Carcinogenesis
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire

Wikimedia Foundation. 2010.