- Prion (proteine)
-
Prion (protéine)
 Pour les articles homonymes, voir prion.
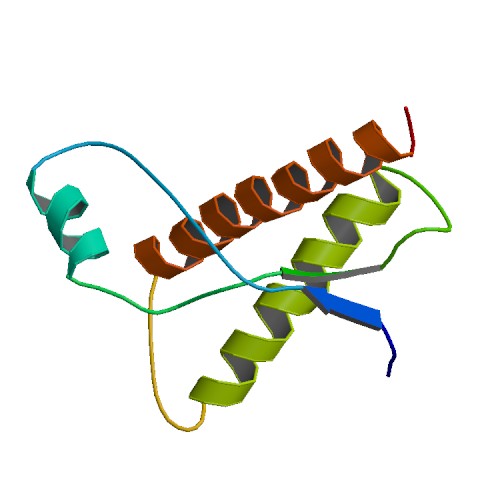
Pour les articles homonymes, voir prion.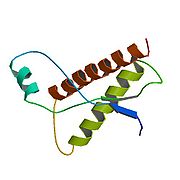 Topologie de la PrPC
Topologie de la PrPC
Un prion est un type d’agent pathogène de nature protéique (constitué d’une protéine ayant adopté une conformation ou un repliement anormal) qui au contraire des agents infectieux conventionnels tels que les virus, les bactéries ou en encore les parasites, est exempt d’acide nucléique (ADN et ARN) comme support de l’information infectieuse. Ce terme fut introduit pour la première fois en 1982 par Stanley Prusiner et correspond à l’acronyme de PROteicaneous INfectious particles (particules protéiques infectieuses).
On distingue les prions de mammifères qui infectent l’homme et différentes espèces animales, des prions retrouvés chez les champignons comme par exemple chez Saccharomyces cerevisiae (levure de boulanger). Les prions de mammifères sont les agents causals responsables des encéphalopathies spongiformes transmissibles (EST) ou maladies à prion. Parmi les EST les plus connues, on peut citer chez l’homme, les différentes formes de la maladie de Creutzfeldt-Jakob, l’insomnie fatale familiale (IFF), le syndrome de Gerstmann-Sträussler-Scheinker (SGSS), le Kuru et chez l’animal, la tremblante du mouton et de la chèvre, l’encéphalopathie spongiforme bovine (ESB), l’encéphalopathie spongiforme féline, l’encéphalopathie spongiforme du vison et le dépérissement chronique du cervidé (CWD pour Chronic Wasting Disease). L’ensemble de ces maladies se caractérise par une dégénérescence du système nerveux central (cerveau et moelle épinière) liée à la propagation ou multiplication des prions chez l’hôte infecté. D'un point de vue anatomo-pathologique, on observe ainsi au niveau de l'encéphale la formation de vacuoles (donnant un aspect spongieux au cerveau, d'ou le nom de spongiforme dans EST), une mort des neurones, une gliose (multiplication des astrocytes et de la microglie) et l'accumulation d'une protéine de l'hôte, la PrPC, sous une conformation anormale (ou mal repliée) alors dénommée PrPSc.
Sommaire
Historique
- 1732 : Première description de la tremblante.
- 1917-1918 : Première description de la maladie de Creutzfeldt-Jakob.
- 1936 : Première description du syndrome de Gerstmann-Sträussler-Scheinker.
- 1938 : Démonstration expérimentale du caractère transmissible de la tremblante. Cuillé et Chelle ont injecté à des moutons des homogénats (tissus broyés) de cerveaux de moutons morts de la tremblante. Les animaux inoculés ont à leur tour déclarés la maladie démontrant ainsi que la tremblante était due à un agent infectieux.
- 1957 : Première description du Kuru par Gajdusek et Zigas.
- 1959 : Hadlow remarque qu'il existe de nombreuses similitudes en termes d'anatomo-pathologie entre la tremblante du mouton et le Kuru. Ces observations le conduisent à émettre l'hypothèse que le kuru pourrait également être causé par un agent infectieux.
- 1966 : Démonstration par le groupe de Gajdusek du caractère transmissible du Kuru au chimpanzé.
- 1967 : Alper ainsi que Pattison réalisent des études d'inactivation (ou destruction) de l'agent infectieux de la tremblante par des rayonnements ionisants. Les rayonnements ionisants conduisent à des altérations et des modifications des acides nucléiques à l'origine de l'inactivation de l'agent pathogène. La dose de rayons ionisants nécessaire pour inactiver la moitié des particules infectieuses est proportionnelle à la taille du génome et donc de la taille de l'agent pathogène lui même. Les résultats qu'ils obtiennent par cette méthode d'estimation de taille, montre que l'agent responsable de la tremblante présente des propriétés de résistance aux rayons ionisants sans commune mesure avec celles obtenues pour les virus, les bactéries et les parasites. Ces observations suggèrent que l'agent infectieux responsable de la tremblante a une taille très petite et bien inférieure à celle des virus ce qui les conduit à formuler l'hypothèse que cet agent infectieux pourrait être dépourvu d'acide nucléique.
- 1967 : Griffith propose l'hypothèse de protéine seule (ou de protein only). Il émet l'hypothèse que l'agent infectieux responsable de la tremblante pourrait être réduit à une protéine ayant adopté un repliement anormal. Cette protéine serait capable d'imprimer sa conformation anormale à la protéine de l'hôte ce qui constituerait le mode de propagation de cet agent.
- 1968 : Le groupe de Gajdusek montre que la maladie de Creutzfeldt-Jakob est également transmissible. Les travaux de Gajdusek permirent ainsi de regrouper les EST sous cette terminologie et t'établir qu'elles étaient dues à un agent infectieux.
- 1978 : Première description du CWD.
- 1982 : Le groupe de Prusiner réalise une étude systématique portant sur l'inactivation de l'agent de la tremblante après transmission au hamster syrien. Ils montrent que tous les traitements physiques et chimiques détruisant les acides nucléiques sont incapables d'inactiver l'agent infectieux. A l'inverse, tous les procédés chimiques et physiques qui détruisent ou détériorent les protéines entraînent une inactivation importante de l'agent infectieux. Prusiner propose d'introduire un nouveau terme pour désigner ce nouveau type d'agent infectieux vraisemblablement dépourvu d'acide nucléique et de nature essentiellement protéique : prion. Ainsi, il remet en cause le paradigme médical de trois sortes d'agents infectieux: Les virus, les microbes et les parasites. La notion de prion est alors mal acceptée par le communauté médicale[1].
- 1982 : Le groupe de Prusiner identifie une protéine qui copurifie avec l'agent infectieux. Ils nommèrent cette protéine PrP, abréviation pour Protease Resistant Protein (protéine résistante aux protéases), car elle présentait la particularité d'être partiellement résistante à la digestion par les protéases (enzymes qui coupent les protéines en petits peptides).
- 1991 : Le groupe de Prusiner établit que la PrP n'est pas une protéine codée par le génome de l'agent infectieux mais par le génome de l'hôte. Ces résultats montrent que chez les individus infectés la PrP normale ou PrPC, exprimée par l'hôte, est convertie (ou subi un repliement) en PrP anormale ou PrPSc.
- 1986 : Première description de l'ESB.
- 1986 : Première description de l'IFF.
- 1993 : Développement des premières souris avec le gène Prnp, codant pour la protéine PrP, invalidé. Ces souris qui n'expriment plus la PrPC ne peuvent plus contracter une maladie à prion après infection expérimentale.
- 1996 : Première description du variant de la maladie de Creutzfeldt-Jakob.
Protéines normales et pathologiques
Le prion ou protéine Prp-sc est une forme spéciale de la protéine Prp-c qui est présente à l’état naturel et est impliquée dans le fonctionnement normal de la cellule. Les fonctions de Prp-c ne sont pas encore connues précisément mais on les soupçonne essentielles. En effet, la protéine Prp-c était présente avant la spéciation des mammifères, ce qui signifie que tous les mammifères (et donc l'homme) sont susceptibles de développer des maladies à prions. La protéine Prp-c est impliquée dans le développement du système nerveux chez l'embryon. Chez l'adulte, elle est exprimée essentiellement dans le cerveau et la moelle épinière (neurones et glie). Elle est impliquée dans les processus de différenciation et d’adhésion des cellules. Elle aurait aussi un rôle protecteur antioxydant et vis-à-vis de la mort cellulaire programmée (apoptose). Cette protéine aurait également un rôle dans le repliement d’autres protéines.
Selon l'équipe du Dr Scott (décembre 2006), la protéine normale, étudiée chez le rat, présente des accumulations particulières à l'intérieur des cellules du pancréas spécialisées dans la production d'insuline, et les rats prédisposés au diabète présentent 3 fois plus de cellules productrices d'insuline avec des amas de protéines Prp-c. Le taux de Prp-c dans le pancréas d'un rat normal change fortement dans les un à trois jours suivant l'administration de concentrations élevées de sucre via le sang. La protéine Prp-c pourrait être impliquée dans le diabète de type 1 ou juvénile, maladies caractérisées par une attaque par le système immunitaire des cellules produisant l'insuline (dans le pancréas)[2].
Le prion est une protéine Prp-c repliée différemment, noté Prp-sc. La Prp-sc résulte d’une modification de la structure tridimensionnelle de Prp-c. Elle provoque les maladies à prions (maladie de la vache folle, ou encéphalopathie spongiforme bovine, maladie de Creutzfeldt-Jakob, tremblante du mouton, Chronical Wasting Disease ou maladie du dépérissement chronique des cervidés). Lors de l'infection, l'agent prion, agent pathogène responsable de l'infection, pénètre le neurone, où pour des raisons et par un mécanisme encore mal compris il se multiplie, en dépliant/repliant les protéines Prp-c en protéines Prp-sc, forme qui n'est plus dégradée par protéolyse et qui, par accumulation dans la cellule, finit par le tuer et former des plaques de dépots dans le cerveau.
Dans toutes ces maladies, aucun acide nucléique (ADN/ARN) n’a pu être spécifiquement associé à l’infectiosité, comme a pu l'être la protéine Prp-sc. On parle d'agent transmissible non conventionnel (ATNC).
Les maladies à prions sont transmissibles d’un individu à l’autre et dans une certaine mesure d’une espèce à l’autre.
Maladies
Troubles dus à sa présence
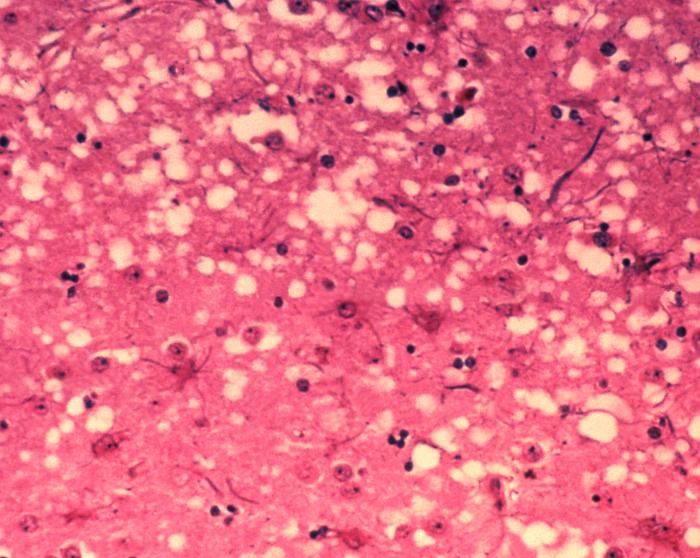
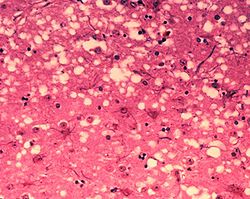 Les « trous » microscopiques sont caractéristiques des tissus infectés de prions, leur donnant une consistance spongieuse.
Les « trous » microscopiques sont caractéristiques des tissus infectés de prions, leur donnant une consistance spongieuse.Les maladies à prions provoquent une dégénérescence du système nerveux central qui est toujours fatale.
- Le rôle de prions est établi dans certaines affections animales telles que l'encéphalopathie spongiforme bovine (ESB ou maladie de la vache folle), la tremblante du mouton et de la chèvre, et la maladie du dépérissement chronique des cervidés par Stanley Prusiner.
- Chez l’homme, il est responsable de la maladie de Creutzfeldt-Jakob qui se caractérise par une démence précoce aboutissant au décès. La forme commune est sporadique, atteignant le plus souvent le sujet âgé. Elle peut être rarement familiale, avec dans ce cas une implication du gène de la protéine prion. Elle peut être également transmise par inoculation de tissus contaminés (extraits d’hypophyse auparavant employés dans le traitement par l’hormone de croissance, greffes de cornée et de dure-mère, électrodes contaminées).
En mars 1996, est apparue une forme clinique chez le sujet jeune (< 30ans), appelé nouveau variant de la maladie de Creutzfeld-Jakob, dont le lien avec l’ESB a été prouvé ensuite. La transmission serait due probablement à l’ingestion de viande bovine contaminée par l’ESB. Le prion est également la cause d’autres maladies humaines : le kuru aujourd’hui disparu (touchant une tribu de Papous de Nouvelle-Guinée qui a été la 1re encéphalopathie spongiforme humaine dont la transmissibilité au singe a été démontrée), la maladie de Gertsmann-Sträussler-Scheinker et l’insomnie fatale familiale.
Il existe d’autres maladies neurologiques comportant des accumulations de protéines anormales, telles la maladie d'Alzheimer et la maladie de Parkinson. La responsabilité d’un prion n’a toutefois pas été démontrée dans ces cas, bien qu’il puisse coexister.
Troubles dus à l’absence de Prp-c
Les données disponibles proviennent d’expérimentation de transgenèse sur des souris/hamsters à qui on a retiré le gène de la protéine Prp-c et qui donc ne possèdent plus cette protéine, ou dont on peut stopper à volonté la production de protéine Prp-c. Ces travaux permettent d’élucider peu à peu les fonctions de la protéine. Certaines souris dépourvues de protéine par knock-out du gène prnp codant cette protéine, sont viables et fertiles, sans phénotype apparent. D'autres développent une mort neuronale massive au niveau du cervelet. Cette mort est due à une autre protéine, paralogue à la protéine saine Prp-c, appelée Doppel (Dpl).
Ce sont plus des modèles expérimentaux que de véritables prions puisqu’il manque dans ces cas la notion d'«infection». Les « Prp-c » de levure ne forment pas des protéines prion comme chez les animaux, mais sont en réalité des protéines (souvent de choc thermique) qui en miment le comportement : dans certaines conditions de stress, elles changent de conformation et s'accumulent, perturbant le fonctionnement cellulaire de la levure.
Mécanismes
Quand la machinerie et les composants nécessaires(ARN-polymérase, ribosome, etc.) sont présents, il est possible de fabriquer des protéines à partir de l’ADN conformément au programme qu’il contient. Toutefois, à composition identique, une protéine peut posséder plus d’une façon de se replier, soit des conformations différentes.
On a constaté que la protéine prion anormale favorise un type de repliement anormal. Or de la bonne ou de la mauvaise façon dont est repliée une protéine dépend sa fonctionnalité.
Le plus puissant ordinateur du monde (en 2004), Blue gene, a été commandé par le Lawrence Livermore Laboratory pour étudier de façon systématique, par simulation, les repliements de protéines en présence et en l’absence de prions.
La levure de bière pourrait être un modèle expérimental intéressant : certaines de ses protéines ont des propriétés de « contagion de forme » qui évoquent celles des prions, même si l’assimilation à ces dernières est discutée.
Détection
Le diagnostic de la maladie est fait de manière courante chez l’animal mort, sur des prélèvements de tissus neurologiques, dans lesquels la concentration en protéine prion anormale est la plus importante chez l’individu malade. Les tests se basent sur l'analyse des résidus de la protéine après digestion à la protéase et également sur la détection de la protéine prion anormale à l’aide d’anticorps spécifiques. Deux méthodes sont utilisées afin de localiser la protéine dans les tissus : le western-blot qui permet l'analyse de la taille des résidus après digestion protéolytique et l'immuno-histo-chimie (ou immunohistochimie) qui met en évidence le complexe anti-corps spécifique/ protéine prion anormale.
En gagnant en sensibilité (détection d’un faible nombre de particules), on espère pouvoir faire, dans l’avenir, un diagnostic par une simple prise de sang sur un sujet vivant.
On peut également rechercher le prion dans d’autres organes, en particulier dans les muscles.
Traitements
Il n'existe aucun traitement à l'heure actuelle.
Préventif
Il repose sur :
- la détection et l’élimination des animaux porteurs ;
- la détection des sujets à risque devant conduire à des précautions accrues lorsqu’ils nécessitent une exploration.
Un vaccin est difficile à trouver du fait de la présence de la protéine normale dans l'organisme. Des chercheurs helvétiques ont donc modifié les gènes des souris pour que leurs lymphocytes B fabriquent des anticorps qui sauront différencier un Prp-sc d'un Prp-c normal. Néanmoins, il n’existe à ce jour pas de vaccin, ni de sérum ayant démontré une efficacité.
Curatif
Plusieurs molécules ont été testées et semblent avoir montré un ralentissement de la progression de la maladie[Laquelle ?]. Parmi elles on peut citer la quinacrine, un anti-paludéen, et le polysulfate de Pentosan.[réf. nécessaire]
Les principaux obstacles à un traitement efficace est qu’il s’agit de maladies de l’encéphale, séparé de la circulation sanguine par une barrière hémato-encéphalique empêchant le passage de la plupart des molécules et que le système immunitaire ne reconnaît pas ce type d'agents infectieux.
Éradication
Le prion est une protéine solide, détruite essentiellement par les hautes températures (autoclave à 134°C pendant 18 min[3]). Il existe également des méthodes chimiques telles que l'eau de Javel fraîchement diluée à 6° chlorométrique et la soude utilisées à température ambiante pendant 1 heure. Ne possédant pas de métabolisme, il n’est guère vulnérable aux irradiations utilisées habituellement dans un but de stérilisation. Cependant, aucune de ces méthodes n’offre une garantie absolue ; l’efficacité maximale est obtenue en associant un traitement chimique au traitement thermique. Les déchets inactivés par ces méthodes doivent ensuite être incinérés dans un centre agréé.
En 2004, l'Institut de génétique humaine (IGH), à Montpellier, a déposé un brevet pour leur découverte de la dégradation par l'action combinée du cuivre et d'un agent oxydant comme l'eau oxygénée [4].
Divers
La recherche sur le prion a fait l’objet de deux prix Nobel de médecine :
- D. Carleton Gajdusek en 1976 pour ses travaux sur le kuru ;
- Stanley B. Prusiner en 1997 pour sa théorie sur le prion, protéine infectieuse.
Bibliographie
Corinne Ida Lasmézas, Qu'est-ce qu'un prion ?, Le Pommier, collection : Les Petites Pommes du Savoir n°65, (ISBN 2746502232)
Notes et références
- ↑ L'histoire du prion sur infodoc.inserm.fr
- ↑ Institut de recherche sur en santé d'Ottawa
- ↑ Association française de stérilisation
- ↑ Une nouvelle arme contre les prions. Consulté le 24 juillet 2009.
Voir aussi
Liens internes
Liens externes
- (fr) Apports de la biologie et de la génétique pour la compréhension de l’évolution
- (fr) Qu'est-ce qu'un prion?
 Portail de la biochimie
Portail de la biochimie
Catégories : Prion | Neurophysiologie
Wikimedia Foundation. 2010.