- Potentiels thermodynamiques
-
Potentiel thermodynamique
Potentiels thermodynamiques Énergie interne U(S,V,N) Énergie libre F(T,V,N) = U − TS Enthalpie H(S,p,N) = U + pV Enthalpie libre G(T,p,N) = U + pV − TS modifier Un potentiel thermodynamique est une fonction associée à un système thermodynamique dans des conditions données (par exemple : pression constante, température constante etc.). Elle a la dimension d'une énergie. De manière analogue à l'énergie potentielle en mécanique, un potentiel thermodynamique représente la quantité d'énergie potentielle du système et est minimal à l'équilibre. De plus, il est possible de déduire d'un potentiel thermodynamique toutes les propriétés du système à l'équilibre par dérivation successives. Différents potentiels thermodynamiques correspondent aux différentes conditions auxquelles peuvent être soumis les systèmes thermodynamiques.
Les quatre potentiels thermodynamiques les plus couramment utilisés sont :
Nom Formule Variables naturelles Énergie interne 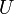
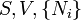
Énergie libre de Helmholtz 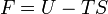
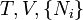
Enthalpie 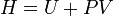
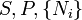
Enthalpie libre de Gibbs 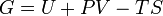
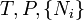
où T = température, S = entropie, P = pression, V = volume. L'énergie libre de Helmholtz est souvent notée F, mais l'utilisation de A est conseillée par l'Union internationale de chimie pure et appliquée. Ni est le nombre de particules de type i dans le système. On a inclus ici l'ensemble des Ni parmi les variables naturelles des potentiels thermodynamiques, mais il est souvent ignoré.
Sommaire
Description et interprétation
Les potentiels thermodynamiques sont très utiles pour calculer le résultat d'équilibre d'une réaction chimique ou bien pour mesurer les propriétés des espèces lors d'une réaction chimique. Les réactions chimiques ont en général lieu sous certaines contraintes simples comme à pression et température constantes, ou bien à entropie et volume constants; dans ce cas-là, on peut faire intervenir les potentiels thermodynamiques correspondants. Comme en mécanique, le système verra la valeur du potentiel diminuer, et à l'équilibre, sous ces contraintes, le potentiel va adopter une valeur minimale permanente. Les potentiels thermodynamiques peuvent également être utilisés pour estimer la quantité totale d'énergie disponible pour un certain système thermodynamique sous certaines contraintes.
En particulier (voir le principe de l'énergie minimale pour la dérivation) :
- Quand l'entropie (S) et les paramètres extensifs (par exemple le volume) d'un système fermé sont maintenus constants, l'énergie interne (U) diminue et atteint un minimum à l'équilibre. C'est une conséquence du premier et du deuxième principe de la thermodynamique, appelée principe de l'énergie minimale. Les trois énoncés ci-dessous sont directement déductibles de ce principe.
- Quand la température (T) et les paramètres extensifs d'un système fermé sont maintenus constants, l'énergie libre de Helmholtz (A) diminue et atteint un minimum à l'équilibre.
- Quand la pression (P) et les paramètres extensifs d'un système fermé sont maintenus constants, l'enthalpie (H) diminue et atteint un minimum à l'équilibre.
- Quand la température (T), la pression (P) et les paramètres extensifs d'un système fermé sont maintenus constants, l'enthalpie libre de Gibbs (G) diminue et atteint un minimum à l'équilibre.
Variables naturelles
Les variables maintenues constantes sont appelées variables naturelles de ce potentiel. Les variables naturelles sont importantes dans le raisonnement ci-dessus, mais également pour la raison suivante : si un potentiel thermodynamique peut être déterminé comme fonction de ses variables naturelles, toutes les propriétés thermodynamiques du système peuvent être déterminées en prenant les dérivées partielles de ce potentiel par rapport à ces variables naturelles (et ce n'est vrai pour aucune autre combinaison de variables). Réciproquement, si un potentiel thermodynamique n'est pas donné comme une fonction de ses variables naturelles, il ne pourra en général pas décrire toutes les propriétés thermodynamiques du système.
Variables conjuguées
Une petite augmentation d'énergie dans un système mécanique est le résultat d'une force multipliée par un petit déplacement. Ainsi une augmentation de l'énergie d'un système thermodynamique peut être exprimé comme une somme de produits de certaines "forces" généralisées, qui hors équilibre provoquent des "déplacements" généralisés. Le produit de cette "force" par ce "déplacement" représente l'énergie transférée. Ces forces et leurs déplacement associés sont appelés variables conjuguées.
Par exemple, considérons la paire conjuguée PV. La pression P agit comme une force généralisée : une différence de pression force un changement dans le volume dV, et leur produit est l'énergie perdue par travail par le système. Ici la pression est la force et le volume associé le déplacement, et les deux variables forment une paire de variables conjuguées. De même, les différences de température induisent des variations de l'entropie, et leur produit est l'énergie transférée par transfert de chaleur.
La force thermodynamique est toujours une variable intensive, et le déplacement une variable extensive. La variable intensive est la dérivée de l'énergie interne selon la variable extensive, avec toutes les autres variables extensives maintenues constantes.
D'autres variables conjuguées - le potentiel chimique
La théorie des potentiels thermodynamiques n'est pas complète tant que l'on ne considère pas le nombre de particules d'un système comme une variable à part entière comme les autres grandeurs extensives telles que le volume ou l'entropie.
Le nombre de particules est la variables de déplacement d'une paire de variables conjuguées. La force généralisée de cette paire est le potentiel chimique. Le potentiel chimique peut être vu comme une force qui, hors équilibre, induit des échanges de particules, soit avec l'extérieur, soit entre les phases d'un système. C'est un concept utile dans les cas de mélanges d'espèces et de phases.
Par exemple, pour un certain volume contenant de l'eau liquide et de la vapeur d'eau, il existe un potentiel chimique (négatif) pour le liquide qui pousse les molécules d'eau vers la vapeur (évaporation), et un potentiel chimique pour la vapeur, qui pousse les molécules de vapeur dans l'eau (condensation). Ce n'est que quand ces "forces" s'équilibrent et que les potentiels chimiques de chaque phase sont égaux que l'équilibre est atteint.
D'autres potentiels thermodynamiques
L'ensemble de variables naturelles pour les quatre potentiels thermodynamiques décrits ci-dessus sont formés de toutes les combinaisons de T-S et P-V, tant que l'on n'utilise pas deux variables conjuguées pour un même potentiel. Il n'y a pas de raison d'ignorer la paire conjuguée µi-Ni, et l'on peut donc définir quatre potentiels thermodynamiques additionels pour chaque espèce. En utilisant les notations de l'UICPA, pour lesquelles les crochets contiennent les variables naturelles autres que les quatre principales, on obtient :
Formule Variables naturelles ![U[\mu_j]=U-\mu_jN_j\,](/pictures/frwiki/50/269912f4aa53f453130defff4762ac97.png)
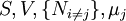
![A[\mu_j]=U-TS-\mu_jN_j\,](/pictures/frwiki/102/fbf99160142ff7ad27d5bc26816c2fdb.png)
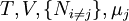
![H[\mu_j]=U+PV-\mu_jN_j\,](/pictures/frwiki/53/5b1c9d3955948331e446f094dd0bf5e7.png)
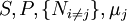
![G[\mu_j]=U+PV-TS-\mu_jN_j\,](/pictures/frwiki/52/447d4f512a5c09213e4f4bceea569229.png)
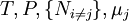
S'il y a par exemple deux espèces, il y aura des potentiels additionels comme U[µ1,µ2] = U - µ1N1 - µ2N2 etc... S'il y a D dimensions dans l'espace thermodynamique, il y aura 2D potentiels thermodynamiques uniques. Pour le cas le plus simple, un gas idéal à phase unique, il y a trois dimensions, et donc huit potentiels thermodynamiques.
Équations fondamentales
Les définitions des potentiels thermodynamiques peuvent être différentiées, et l'on peut obtenir un ensemble d'équations différentielles connues sous le nom d'« équations fondamentales ».
D'après le premier principe de la thermodynamique, une variation différentielle de l'énergie interne U d'un système peut s'écrire comme la somme d'une quantité de chaleur fournie au système, de travail fourni à l'environnement par le système, et d'un terme dû à l'apport de nouvelles particules au système :
où δQ est la quantité infinitésimale de chaleur fournie au système, δW est le travail infinitésimal fourni par le système, µi est le potentiel chimique des particules de type i, et Ni est le nombre de particules de type i (ni δQ ni δW sont des différentielles exactes. Des petites variations de ces grandeurs sont donc représentées par δ plutôt que par d).
D'après le second principe de la thermodynamique, on peut exprimer les variations d'énergie interne grâce à des fonctions d'état et leurs différentielles :
où T est la température, S l'entropie, P la pression et V le volume (l'égalité est obtenue pour les transformations réversibles).
Ceci nous mène à la forme différentielle standard de l'énergie interne :
En appliquant la transformation de Legendre, les relations différentielles pour les autres potentiels thermodynamiques principaux sont donc :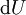
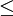
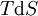

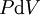
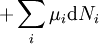
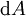
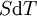
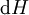
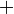
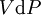
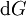
Pour chaque équation, les différentielles du membre de droite sont les variables naturelles du potentiel décrit au membre de gauche. Ces relations montrent que quand les variables naturelles de chaque potentiel sont maintenues constantes, la valeur du potentiel diminue de manière irréversible, vers sa valeur minimale constante à l'équilibre.
Des équations similaires peuvent être développée pour les autres potentiels thermodynamiques du système. Il y a une équation fondamentale pour chaque potentiel thermodynamique, soit au final 2D équations fondamentales.
Equations d'état
On peut utiliser les équations ci-dessus pour en dériver les définitions différentielles de quelques grandeurs thermodynamiques. Si l'on note Φ un potentiel thermodynamique quelconque, alors les équations précédentes sont de la forme :
où xi et yi sont des paires conjuguées, et les yi sont les variables naturelles du potentiel Φ. D'après la règle de dérivation en chaîne :
où
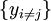 est l'ensemble de toutes les variables naturelles de Φ excepté yi. Ceci nous amène aux expression des grandeurs thermodynamiques comme dérivée partielles de potentiels selon leurs variables naturelles. Ces équations sont connues sous le nom d' équations d'état, étant donné qu'elles décrivent les paramètres de l'état thermodynamique du système. En se limitant aux quatre potentiels principaux U, A, H et G, on a :
est l'ensemble de toutes les variables naturelles de Φ excepté yi. Ceci nous amène aux expression des grandeurs thermodynamiques comme dérivée partielles de potentiels selon leurs variables naturelles. Ces équations sont connues sous le nom d' équations d'état, étant donné qu'elles décrivent les paramètres de l'état thermodynamique du système. En se limitant aux quatre potentiels principaux U, A, H et G, on a :où, dans la dernière équation, φ est un potentiel thermodynamique parmi U, A, H et G, et
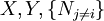 sont les ensembles de variables naturelles pour ce potentiel, excepté Ni. Si l'on souhaite décrire tous les potentiels, on obtient des équations d'état supplémentaires telle que
sont les ensembles de variables naturelles pour ce potentiel, excepté Ni. Si l'on souhaite décrire tous les potentiels, on obtient des équations d'état supplémentaires telle queetc... Au total, il y a D équations pour chaque potentiel, soit D×2D équations d'état. Si les D équations d'état pour un potentiel particulier sont connues, alors l'équation fondamentale pour ce potentiel peut être déterminée. Ceci signifie que toutes les informations thermodynamiques sur le système peuvent être connues et que les équations fondamentales de chaque potentiel peuvent être déterminées.
Relations de Maxwell
A nouveau, on définit xi et yi comme des variables conjuguées et yi comme les variables naturelles d'un potentiel Φ. En prenant les "différentielles croisées" des équations d'état, qui obéissent à la relation :
on obtient les relations de Maxwell. Il y a (D-1)/2 relations pour chaque potentiel, soit un total de D(D-1)/2 équations au total. En se limitant à U, A, H, et G, on a :
En utilisant les équations d'état incluant le potentiel chimique, on obtient des équations telles que :
et en utilisant les autres potentiels, on obtient des équations telles que :
Intégrales d'Euler
A nouveau, on définit xi et yi comme des paires conjuguées, et yi comme les variables naturelles de l'énergie interne. Comme toutes les variables naturelles de l'énergie interne U sont des grandeurs extensives :
d'après le Théorème d'Euler pour les fonctions de plusieurs variables, l'énergie interne peut être écrite sous la forme :
L'équation d'état nous donne :
Par substitution dans les expressions des autres potentiels thermodynamiques, on obtient :
Comme dans les sections précédentes, on peut effectuer cette suite de calculs avec tous les autres potentiels thermodynamiques. Les intégrales d'Euler sont parfois appelées équations fondamentales.
La relation de Gibbs-Duhem
En différentiant l'équation d'Euler pour l'énergie interne ci-dessus, on a :
Or l'équation fondamentale pour U énonce que :
Par soustraction, on obtient la relation de Gibbs-Duhem :
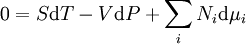
La relation de Gibbs-Duhem, établie par Josiah Gibbs et Pierre Duhem, est une relation entre les paramètres intensifs du système. Il en découle que pour un système simple à r composants, il y aura r+1 paramètres indépendants, ou degrés de liberté. Par exemple, un système simple avec un composant unique a deux degrés de liberté, et peut être décrit par deux paramètres, comme la pression et le volume par exemple.Réactions chimiques
Les variations de ces grandeurs sont utiles pour décrire le degré d'avancement d'une réaction chimique. La grandeur appropriée dépend des conditions de réaction, comme le montre le tableau suivant :
V constant P constante S constante ΔU ΔH T constante ΔA ΔG On considère souvent les réactions à P et T constantes, c'est donc l'enthalpie libre de Gibbs qui sera le potentiel le plus utile pour étudier la réaction chimique.
Références de l'article anglais
- Alberty, R. A., « Use of Legendre transforms in chemical thermodynamics », dans Pure Appl. Chem., vol. Vol. 73, no 8, 2001, p. 1349–1380 [texte intégral]
- Callen, Herbert B. (1985). Thermodynamics and an Introduction to Themostatistics, 2nd Ed., New York: John Wiley & Sons. ISBN 0-471-86256-8.
Voir aussi
- Fonction caractéristique
- Équilibre canonique
- Potentiel chimique
- Variables conjuguées
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « Thermodynamic potentials ».
 Portail de la physique
Portail de la physique
Catégorie : Thermodynamique
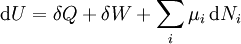
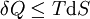
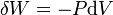
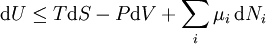
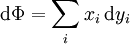
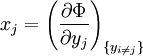
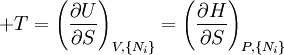
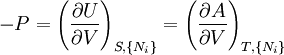
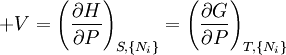
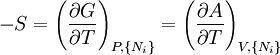
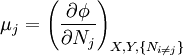
![-N_j=\left(\frac{\partial U[\mu_j]}{\partial \mu_j}\right)_{S,V,\{N_{i\ne j}\}}](/pictures/frwiki/51/3c18bf0022d4ea560cad992fb8ec60e5.png)
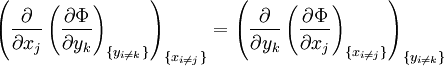
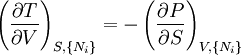
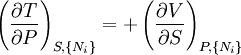
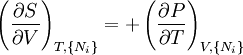
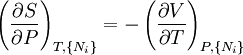
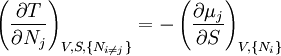
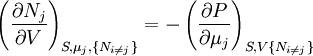
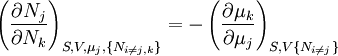
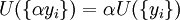
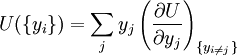
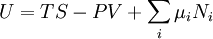
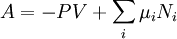
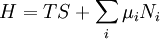
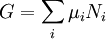
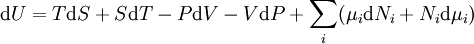
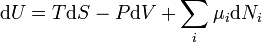
Wikimedia Foundation. 2010.