- Organo-magnésien
-
Organomagnésien
Les organomagnésiens sont des composés organiques possédant une liaison carbone–magnésium, ils font partie de la famille des organométalliques.
C'est en 1912 que Victor Grignard, chimiste français, reçoit le Prix Nobel de chimie pour la synthèse et les applications des organomagnésiens mixtes — les premiers organométalliques étudiés, en 1900, par Grignard. Les organomagnésiens sont d'ailleurs aussi appelés « Réactifs de Grignard ».
Les organomagnésiens mixtes ont pour structure commune : R—Mg—X, avec R la chaîne carbonée, Mg l'atome de magnésium, et X un atome halogène — principalement Cl, Br, ou I, plus rarement F de par la faible polarisabilité du difluor F2.
Les organomagnésiens servent d'intermédiaires très utiles dans la fabrication d'autres composés : ils servent dans l'industrie pharmaceutique, mais aussi dans la synthèse des alcools et surtout dans l'élaboration du silicone.
Sommaire
Caractère organométallique des R—Mg—X
Le carbone a une électronégativité χ forte : χC = 2,55. Et on remarque aussi que
 . Ce fait physique impose donc la polarisation de la liaison C—M :
. Ce fait physique impose donc la polarisation de la liaison C—M : 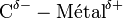 . On remarque une inversion de la polarité des liaisons classiques du carbone : dans un organométallique, le carbone porte une charge δ − , ce qui permettra la création de liaisons C—C.
. On remarque une inversion de la polarité des liaisons classiques du carbone : dans un organométallique, le carbone porte une charge δ − , ce qui permettra la création de liaisons C—C.Les organomagnésiens sont une des bases au sens de Brönsted les plus fortes de la chimie organique avec un pKa de l'ordre de 40 à 60. R—Mg—X est un nucléophile puissant.
Synthèse des organomagnésiens
Du fait de leur extrême basicité, les organomagnésiens n'existent pas à l'état naturel. Le bilan global de la synthèse est le suivant:
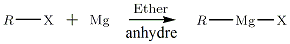
Mode opératoire
La synthèse des organomagnésiens est délicate : le milieu doit être anhydre, en raison des propriétés du produit souhaité ; la réaction est fortement exothermique et le solvant est volatil et inflammable. Une mauvaise manipulation entraîne aussi la création d'un produit parasite — appelé « Couplage de Wurtz ».
Matériel requis :
- Ballon tricol ;
- Ampoule de coulée isobare ;
- Colonne à reflux (de Vigreux);
- tulipe deshydratante contenant CaCl2 (ou bien réaction à effectuer sous atmosphère inerte, N2 par exemple);
- Agitateur mécanique ;
- Copeaux de magnésium ;
- Éther anhydre ;
- Solution de dérivé halogéné R—X en éther anhydre ;
- Bain marie puis eau froide.
Grâce à l'ampoule de coulée, on verse de façon très lente — quelques gouttes par seconde au maximum — la solution de R—X sur l'éther anhydre dans lequel baignent les copeaux de magnésium. Et ce pour éviter la réaction de Würtz entre l'organomagnésien synthétisé et le réactif halogéné. Dès le début de la réaction, l'éther entre en ébullition : la colonne à reflux a pour but de ramener les vapeurs en phase liquide dans le ballon ; le reflux est lui aussi à maintenir très faible, afin de ne pas créer le produit parasite cité précédemment.
La solution finalement obtenue est de couleur marron.
Solvant
On utilise un éther anhydre en raison de leurs propriétés basiques de Lewis. Les doublets non liants de l'oxygène vont permettre de stabiliser le centre réactif du magnésium. On peut par exemple utiliser le diéthyl éther, ou encore le THF — tétrahydrofurane, cf. figure ci-dessous.
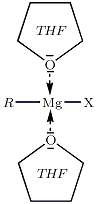
Le milieu dans lequel baigne l'organomagnésien doit être dépourvu d'eau, et ceci de façon scrupuleuse : la réaction eau / RMgX est vive.
Réactivité des organomagnésiens
Réactions sur les composés organiques habituels
Sur l'eau
La réaction des organomagnésiens sur l'eau est très vive (c'est une réaction acido-basique:pKa(RH/R-)=50), et exothermique. C'est pour cela que le mode opératoire impose une deshydratation totale de l'air dans la verrerie, ainsi que des surfaces et solvants. Sauf cas particuliers, cette réaction est considérée comme parasite, et entraine la destruction de l'organomagnésien.
La réaction de l'organomagnésien sur l'eau est la suivante :

Avec les alcools
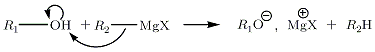
Avec les acides carboxyliques
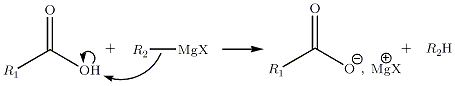
mécanisme faux
Avec les amines
Fichier.gif Avec les alcynes vrais
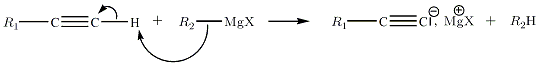
Réactions nucléophiles
Substitution nucléophile, exemple de I2
On propose la substitution nucléophile de RMgX sur I2 (la polaritié de cette molécule est induite).
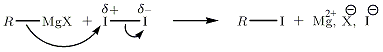
Substitution nucléophile avec les dérivés halogénés
Dérivé halogéné: R'-X mécanisme: SN
R-MgX + R'-X —> R-R' + MgX2
Remarque: fonctionne bien avec R-I et Ar-CH2X car ils sont stabilisés par effet inductif pour R-I et mésomère pour Ar-CH2X
Réaction sur les époxydes
mécanisme: SN2
Dans l'ether:
R----Mg----X + O(CH2)2 (cycle) ---> R-CH2-CH2-O-Mg-X
Puis Hydrolyse acide H3O+ :R-CH2-CH2-O-Mg-X + H+ ---> R-CH2-CH2-OH + MgX
Réactions d'addition nucléophile…
… sur les aldéhydes et cétones
- Sur les aldéhydes :
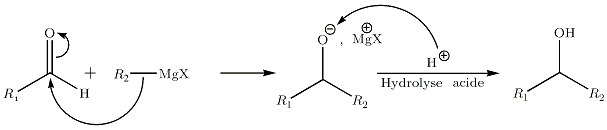
La réaction entre les organomagnésiens et les aldéhydes mènent à la formation d'un alcool secondaire, si les chaines R1 et R2 contiennent au minimum un carbone. Si le carbone d'appui du groupement alcool final est asymétrique, c’est-à-dire si les deux chaines R sont différentes, le produit sera composé des deux isomères de configuration R et S.
- Sur les cétones :
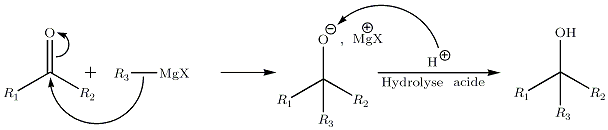
Cette réaction conduit à la création d'un alcool tertiaire. Comme pour les aldéhydes, si le carbone formé est asymétrique, on obtient un mélange R/S 50/50 ; ainsi, il n'y a pas de stéréoselectivité.
- Cas particulier de la synthèse d'un alcool primaire :
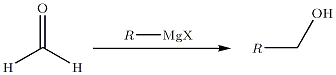
… sur les esters
La réaction est similaire à la réaction sur une cétone. Après hydrolyse, nous obtenons de manière intermédiaire l'hémiacétal;
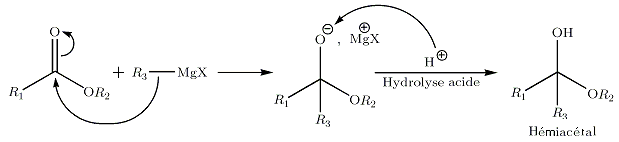
Celui-ci est instable, il subit une élimination conduisant à une cétone. L'organomagnésien présent va réagir chimiosélectivement avec la cétone avant l'ester, moins réactif. Le bilan global de la réaction conduit donc à la formation d'un alcool tertiaire, sans possibilité d'isoler la cétone intermédiaire.… sur le CO2
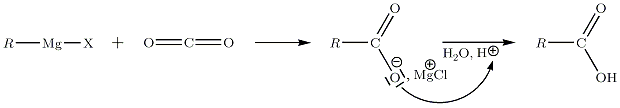
On utilise le dioxyde de carbone à l'état de carboglace. La réaction conduit à la formation d'un acide carboxylique R-COOH.
 Portail de la chimie
Portail de la chimie
Catégories : Groupe fonctionnel | Organomagnésien
Wikimedia Foundation. 2010.