- Phéochromocytome
-
Phéochromocytome
Classification et ressources externes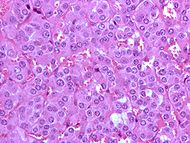
Micrographie d'un phéochromocytome montrant l'atteinte des cellules chromaffines de la médullo-surrénale. CIM-10 C74.1 CIM-9 255.6 ICD-O: M8700/0 OMIM 171300 DiseasesDB 9912 MedlinePlus 000340 eMedicine med/1816 radio/552ped/1788 MeSH D010673 Un phéochromocytome est une affection tumorale parfois maligne. Cette tumeur se développe à partir des cellules chromaffines de la médullo-surrénale. Ne pas confondre avec le corticosurrénalome qui quant à lui se développe à partir du cortex de la glande surrénale.
Cette tumeur est exceptionnelle, mais touche des patients relativement jeunes (20-50 ans). Son caractère malin est encore plus rare, mais redoutable, avec l'émission de métastases hépatiques et osseuses.
Sommaire
Historique
Sa première description remonte à 1886 par Félix Fränkel[1] chez une patiente décédée de ce qu'il pensait être un cancer des glandes surrénales. Le terme phéochromocytome apparaît en 1912[2] et correspond à une coloration particulière des tissus surrénaliens après fixation au chromate.
Diagnostic
Il doit être évoqué devant les symptômes, l'imagerie retrouve une masse surrénalienne. Dans 10 % des cas on retrouve une masse extra-surrénalienne, on parle alors de paragangliome. Le diagnostic définitif est anatomo-pathologique.
Clinique
Les signes cliniques du phéochromocytome sont induits par la sécrétion excessive des catécholamines (adrénaline et noradrénaline). Ce tableau clinique comprend :
- une hypertension artérielle et une perte de poids
associée à une triade symptomatique typique mais inconstante associant :
- des céphalées pulsatiles
- des palpitations cardiaques et tachycardie
- des sueurs profuses
Le phéochromocytome est le plus souvent d'origine génétique, il est donc important de rechercher les éléments qui pourront faire suspecter une maladie de Von Hippel-Lindau, une néoplasie endocrinienne multiple, ou une neurofibromatose de type 1 ou maladie de Recklinghausen
Biologie
- Dosage des catécholamines et de leurs métabolites plasmatiques et urinaires :
- métanéphrines et chromogranine plasmatique
- métanéphrine et normétanéphrine urinaires des 24h
Anomalies génétiques associées
- Recherche d'une néoplasie endocrinienne multiple de type 2 (NEM2) :
- recherche d'une mutation du proto-oncogène RET
- recherche d'une hyperparathyroïdie
- recherche d'un cancer médullaire de la thyroïde
- Recherche d'une mutation du gène VHL (maladie de Von Hippel-Lindau)
- Neurofibromatose de type 1
- Mutation des gènes SDHB, SDHC, SDHD, SDHAF2 ou TMEM127
Imagerie
- Scintigraphie au MIBG (Méta-iodobenzylguanidine)
- Scanner (Tdm) ou IRM thoraco-abdominal
Traitement
La prise en charge du phéochromocytome relève de services hautement spécialisés de fait de :
- la rareté de cette maladie
- la nécessité d'une exploration génétique
- la difficulté de la prise en charge chirurgicale, et en particulier l'anesthésie per-opératoire
- la complexité de la stratégie thérapeutique des formes malignes.
Chirurgical
Ablation de la surrénale (Surrénalectomie) par voie cœlioscopique en général. Chirurgie délicate au vu du risque de poussée hypertensive durant l'intervention. Une préparation médicamenteuse par alpha et bêta bloquants permet de prévenir ce risque.
Notes et références
- Fränkel F, Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat und Retinitis, Arch Pathol Anat Physiol Klin Med, 1886;103:244-63
- Pick L, Das Ganglioma embryonale sympathicum (Sympathoma embryonale), Berl Klin Wschr 1912;49:16-22
Liens externes
Catégories :- Maladie tumorale du système endocrinien
- Terme médical
Wikimedia Foundation. 2010.