- Paralysie supranucleaire progressive
-
Paralysie supranucléaire progressive
 Pour les articles homonymes, voir PSP.
Pour les articles homonymes, voir PSP.La paralysie supranucléaire progressive (PSP ou maladie de Steele-Richardson-Olszewski), en raison de l'ophtalmoplégie supranucléaire qui la caractérise, a été décrite pour la première fois, estime-t-on, en 1904, bien que des recherches d’antériorité évoquent des cas de PSP probable au moins à la fin du 19ième siècle. Ce n’est pourtant qu’en 1964 que la PSP sera identifiée comme une maladie à part entière, et donc comme une entité nosologique spécifique par les trois Professeurs neurologues Steele, Richardson et Olszewski, qui lui ont donné leurs noms. La PSP, paralysie supranucléaire progressive, est donc à ce titre appelée aussi maladie de Steele, Richardson et Olszewski.
Elle affecte aujourd'hui plusieurs dizaines de milliers de personnes aux États-Unis et en Europe (40 à 60 000, selon les estimations). La PSP provoque des lésions du tronc cérébral affectant progressivement l’équilibre, la vue, la mobilité, la déglutition et la parole. La PSP demeure toutefois méconnue et quand elle se déclare, elle est encore mal diagnostiquée avec pour conséquence des traitements initiaux souvent mal adaptés. Ainsi, la plupart des patients atteints de PSP rapportent que leur médecin généraliste ignorait tout de cette maladie jusqu'à ce que le diagnostic soit fait par un neurologue et avec l'aide d'examens par IRM (atrophie Mésencéphalique, élargissement du V4, hypersignal T2 pallidum interne, discret hypersignal T2 bordant le putamen).
Sommaire
Une maladie rare et orpheline
La PSP peut être difficile à reconnaître au début car les troubles initiaux sont également présents dans d’autres maladies neurodégénératives, comme pour les plus connues celles de Parkinson et d'Alzheimer, mais aussi comme celles de l'AMS (Atrophie Multi-Systèmatisée), ou la DCB (Dégénérescence Cortico-Basale) qui est très proche de la PSP, au moins en début de maladie. L'errance de diagnostique demeure encore importante, en moyenne plus de 3 ans, avec de fortes disparités. On considère que la PSP représente 5 à 10 % des syndromes parkinsoniens. Aux États-Unis comme en Europe où la maladie est maintenant mieux reconnue, bien que souffrant encore d'une insuffisance de dépistage, on estime à plusieurs dizaines de milliers les personnes qui en seraient atteintes de chaque coté de l’Atlantique. Toute maladie est une maladie de trop mais la PSP fait partie de celle où le nombre de personnes atteintes demeure malgré tout relativement faible, ce qui en fait une maladie rare. C'est là toute la cruauté de cette rareté, car elle est aussi d'autre part une maladie orpheline car à ce jour aucun médicament n'a pu démontrer une efficacité thérapeutique avérée dans le traitement de la PSP. Cette absence de traitement spécifiquement adapté à sa pathologie est, pour le malade et sa famille, la source de bien des désarrois, nécessitant également une prise en charge de celui ou celle qui accompagne et aide le malade.
Symptomatologie
Les symptômes les plus précoces sont un ralentissement intellectuel, des modifications du comportement pouvant se manifester sous forme d'une perte d'intérêt pour les activités journalières ordinaires ou d'une irritabilité et d'une susceptibilité croissantes. Ces troubles comportementaux font penser à un état dépressif. L'élocution change, elle aussi, avec une impression de bégaiement, de nasillement et de difficultés à émettre des phrases compréhensibles. Ainsi ces symptômes donnent une fausse impression de sénilité. Mais le symptôme le plus impressionnant est la perte d'équilibre à la marche, responsable de chutes inexpliquées, la perte d'équilibre s'aggrave au point de rendre la marche difficile voire impossible. A ces troubles de la mobilité viennent s'ajouter des troubles plus insidieux tels que des difficultés visuelles. Ces problèmes visuels résultent d'une incapacité à guider les yeux correctement du fait de la faiblesse ou de la paralysie des muscles qui contrôlent les globes oculaires (diplopie). Enfin comme les troubles de la vision, les troubles de la déglutition font partie des symptômes plus tardifs de la maladie, ceux-ci pouvant entraîner des « fausses routes » alimentaires (l'asphyxie par étouffement est une fréquente cause de décès en fin d'évolution). Tous ces symptômes découlent d'une paralysie ciblant de façon privilégiée les muscles opérant dans le plan sagittal.
Diagnostic différentiel
Le diagnostic est difficile, surtout en début de maladie. La PSP (paralysie supranucléaire progressive) peut être difficile à différencier de la DCB (dégénérescence cortico-basale) ou d'une AMS (atrophie multi-systématisée). Dans le cas de la DCB, des médecins expérimentés se trompent dans plus de 50% des cas[1].
La PSP, comme la DCB qui lui est très similaire, sont des maladies de mauvais pronostic, avec une évolution par paliers, très invalidante et dont seuls les symptômes peuvent à ce jour être traités. Des soins palliatifs sont à prévoir.
Les diagnostics différentiels principaux sont :
- Maladie de Parkinson
- Dégénérescence Cortico-Basale (DCB)
- Atrophie Multi-Systématisée (AMS)
- Dégénérescence Fronto-Temporale (DFT) dont la Maladie de Pick
Les maladies d'Alzheimer, AMS, DCB, PSP, DFT dont Pick sont des tauopathies.
Depuis mai 2007, dans le cadre du Plan National Maladies Rares[2], en liaison avec l'association PSP France[3], un centre de référence a été labellisé pour la paralysie supranucléaire progressive au sein de l'hôpital de la Salpétrière à Paris.Depuis mars 2009, le Réseau National des Centres de Référence et de Compétence est en place pour la PSP afin d'apporter un supplément d'expertise dans la prise en charge, le traitement et la recherche. L'association PSP France est à ce jour la seule association de malades présente au sein de ce réseau pour les démences rares (classement nosologique).
Perspectives thérapeutiques
Les traitements actuels sont essentiellement symptomatiques et cherchent à lutter contre la gêne occasionnée par la maladie. A ce titre la famille et l'entourage du patient sont essentiels, mais ils doivent eux-mêmes être soutenus et aidés dans cette lutte. La recherche vise à mieux comprendre les mécanismes de la dégénérescence neuronale, afin de ralentir l’évolution de la maladie. Un règlement sur les médicaments orphelins a déjà été adopté par le parlement européen depuis le 15 décembre 1999. Le projet européen NNIPPS, portant sur 800 patients s’est proposé en 2001 d’étudier les spécificités cliniques, cognitives, neuro-radiologiques et histologiques de la PSP. Les résultats définitifs de cette étude ne sont pas encore publiés, mais nous savons hélas, à ce jour, que les essais n'ont pas donné les résultats d'efficacité escomptée. Des recherches sont également réalisées sur la nature des troubles oculomoteurs et de l’équilibre qui sont caractéristiques de l’affection.
La PSP n'est pas héréditaire. Il est mené toutefois des recherches sur d’éventuelles prédispositions afin de déterminer la part d’héritabilité.
Héréditaire s'emploie souvent comme raccourci pour une héritabilité forte, qui peut être de 100 %.
L'héritabilité, elle, est définie comme la part de variance expliquée chez les descendants, dans une analyse de la variance par le facteur génétique (ANOVA) : "une maladie peut être héritable (un ou plusieurs gènes y prédisposent) sans être transmise avec certitude à la descendance".
Enfin de très nombreuses études et recherches sur le cerveau permettent de comprendre de mieux en mieux les mécanismes complexes de son fonctionnement et de son dysfonctionnement. Il y a lieu de penser qu'à terme ces travaux déboucheront sur des thérapies d'autant plus intéressantes qu'elles concerneront tout un ensemble de maladies neurodégénératives et non pas seulement la PSP.
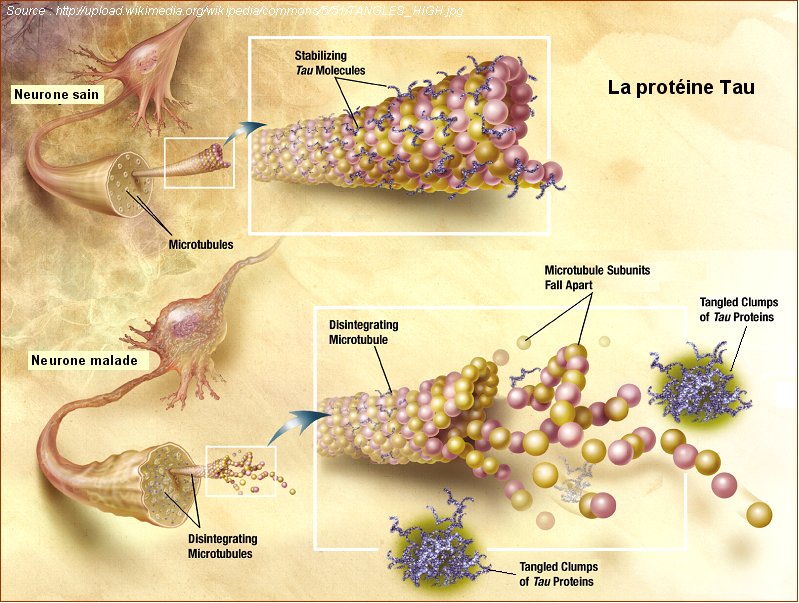
La protéine TAU, une protéine impliquée dans la P.S.P.
Article détaillé : protéine Tau.Le codage des protéines obéit à des mécanismes biochimiques et moléculaires complexes, à dessin d'assumer des fonctions normales de vie ou de mort (apoptose) de nos cellules.
Dans les années 1980, la dégénérescence neurofibrilllaire est associée à la protéine tau qui, longtemps négligée par la recherche, suscite aujourd’hui un regain d’intérêt.
La protéine tau, comme toutes les autres est codée par nos gènes et le gène codant la protéine tau est situé sur le chromosome 17. Tout être humain fabrique de la protéine tau qui est normalement non pathogène et qui même lorsqu'elle peut, avec l'âge, devenir pathogène, elle demeure non toxique car sans expansion dans le système nerveux. Les mécanismes qui président au passage de la situation non pathogène de la protéine Tau à pathogène puis de pathogène à toxique, demeurent encore inexpliqués.
Références
- ↑ Litvan I, and others. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology 1997: 48: 119-125
- ↑ [pdf] Plan National Maladies Rares
- ↑ PSP France
 Portail de la médecine
Portail de la médecine
Catégories : Maladie neurodégénérative | Maladie de l'encéphale
Wikimedia Foundation. 2010.