- Canal Atrio-ventriculaire
-
Canal atrio-ventriculaire
 Pour les articles homonymes, voir CAV.
Pour les articles homonymes, voir CAV.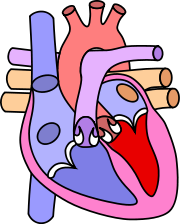 Cœur normal
Cœur normal
Le canal atrio-ventriculaire (CAV) est une malformation cardiaque complexe qui touche la partie basse du septum inter-auriculaire, la partie haute du septum interventriculaire, les valves mitrale et tricuspide. Les valves mitrale et tricuspide sont remplacées par une seule valve, il manque la partie supérieure du septum interventriculaire et la partie inférieure du septum inter-auriculaire. Cette anomalie est un trouble de développement de la crux cordis.
Cette pathologie représenterait 4% des cardiopathies congénitales soit deux C.A.V pour 10 000 naissances. Les deux sexes sont également atteints et c'est la cardiopathie dont le rapport avec les anomalies chromosomiques est la plus forte notamment avec la trisomie 21.
Sommaire
Différents types
Pour la compréhension des différents types, il est souhaitable de lire l'article sur l'embryologie du cœur.
Canal atrio-ventriculaire complet
Canal atrio-ventriculaire incomplet
Diagnostic
Le diagnostic de canal atrio-ventriculaire complet au cours de la grossesse devrait être un objectif de toute échographie de dépistage tant les conséquences sont importantes. Il s'agit d'une des deux cardiopathies (avec la transposition des gros vaisseaux) qui ne devraient pas théoriquement échapper à une analyse morphologique systématique du cœur du fœtus. En effet cette cardiopathie est diagnostiquable sur la coupe dite des quatre cavités : coupe standard qui doit être réalisée au cours de l'examen du fœtus.
Anténatal
Aspects échographiques avant la naissance
Les anomalies de la coupe des quatre cavités dans le canal atrio-ventriculaire complet sont:
- Disparition totale de l'aspect dite en croix de cœur formée par la valve mitrale, le septum interventriculaire et la valve tricuspide qui est plus proche de la pointe du cœur que la valve mitrale.
- La visualisation des deux valves s'ouvrant et se fermant est remplacée par une seule valve auriculo ventriculaire commune.
Les diagnostics à éliminer à ce stade sont une communication inter-ventriculaire importante et/ou communication inter-auriculaire et le ventricule unique. L'aspect peut aussi parfois être confondu avec une tétralogie de Fallot.
Gestion de la grossesse
Cette pathologie aboutit rarement à une défaillance cardiaque in utero avec risque de mort fœtal in utero. En raison de l'association forte avec la trisomie 21, la découverte d'un CAV doit faire conseiller au parent la réalisation d'un caryotype par amniocentèse ou ponction de sang fœtal pour la recherche de la trisomie 21.
Les principaux syndromes chromosomiques et génétiques à rechercher sont:
Nom de la pathologie Autres anomalies à rechercher Transmission Trisomie 21 ++ Sans objet Délétion 8p Syndrome d'Ivemark Syndrome d'Ellis-Van Creveld Polydactylie-Pieds bots Récessive Syndrome de Holt-Oram Anomalie des membres Dominante L'accouchement devra se faire dans un centre disposant d'un centre de réanimation néonatale et d'un service de cardiopédiatrie. Le mode d'accouchement n'est pas affecté par cette pathologie.
Prise en charge du nouveau-né
La présence d'une sténose de l'artère pulmonaire nécessitera la mise en route de prostaglandine pour laisser ouvert le canal artériel. À la naissance, une échocardiographie sera effectuée pour confirmer le diagnostic et rechercher d'autres anomalies non diagnostiquées in utero; le type précis du canal atrio-ventriculaire et l'intégrité des artères coronaires seront déterminés.
Postnatal
Les premiers signes apparaissent généralement dans les deux premières semaines: des difficultés respiratoires, des difficultés à l'alimentation et une hypotonie.
Traitement
Forme partielle
Forme complète
Le traitement chirurgical est impératif en raison du risque précoce d'hypertension artérielle pulmonaire. L'intervention est l'intervention de Rastelli [1].
Le traitement médical (diurétiques et vasodilatateurs) permet d'attendre l'opération qui est pratiquée en général entre trois et six mois de vie.
Le risque opératoire des communications atrioventriculaires partielles est d'environ 3%.
Devenir des enfants
- La survie des patients atteint d'un C.A.V partiel est de 96% à l'âge de 20 ans. Une réintervention est nécessaire dans 9% des cas, pour corriger une régurgitation mitrale ou une sténose sous aortique.[2]
- Sans traitement, la moitié des patients avec un CAV complet décèdent dans la 1er année de vie. La cause principale de décès est l'arrêt cardiaque ou la pneumonie. Chez les patients survivants, non traités, une hypertension pulmonaire apparait et s'aggrave progressivement, touchant tous les patients de plus de deux ans. Le pronostic à long terme de ces patients avec une hypertension pulmonaire irréversible est faible. [3]
Conseil génétique
En l'absence de maladie à transmission héréditaire, le risque d'avoir un autre enfant atteint de cette pathologie est de 3% mais passe à 10% si deux enfants sont atteints[4]. En cas de mère enceinte porteuse de cette pathologie ou d'un père, le risque est de 10% [5].
Sources
- (fr) Alain Batise Cardiologie pédiatrique pratique Doin éditeurs
- (en) Raffaele Calabrò and Giuseppe Limongelli Complete atrioventricular canal Orphanet Journal of Rare Diseases 2006, 1:8 doi:10.1186/1750-1172-1-8
Références
- ↑ GC Rastelli, PA Ongley, JW Kirklin, and DC McGoon Surgical repair of the complete form of persistent common atrioventricular canal J. Thorac. Cardiovasc. Surg. 55: 299-308.
- ↑ Titre
- ↑ Orphanet Journal of Rare Diseases | Full text | Complete atrioventricular canal
- ↑ Nora JJ, Nora AH. Update on counselling the family with a first-degree relative with a congenital heart defect Am J Med Genet 1988;29;137-142
- ↑ Emmanuel R, Sommerville J, Ins A, et al. Evidence of congenital heart disease in the offspring of parents with atrio-ventricular defect. Br Heart J 1983;49:144-147
 Portail de la médecine
Portail de la médecine
Catégories : Médecine fœtale | Cardiopathie congénitale

Wikimedia Foundation. 2010.