- Dystrophie facio scapulo humérale
-
Dystrophie facio-scapulo-humérale
La dystrophie facio scapulo humérale ou myopathie facio-scapulo-humérale ou myopathie de Landouzy-Dejerine est une affection musculaire héréditaire liée à une anomalie située sur le chromosome 4. Actuellement, seule la kinésithérapie est utile comme traitement.
Sommaire
Description clinique de la maladie
 Patiente de 27 ans atteinte d'une forme de FSH concernant préférentiellement la musculature axiale du tronc. On note une hyperlordose non structurelle due au déficit de la musculature paravertébrale lombaire prédominant sur les fléchisseurs du rachis. Le passage en position verticale ne peut se faire qu'en penchant le bassin en avant. On note aussi un décollement bilatéral marqué des omoplates.
Patiente de 27 ans atteinte d'une forme de FSH concernant préférentiellement la musculature axiale du tronc. On note une hyperlordose non structurelle due au déficit de la musculature paravertébrale lombaire prédominant sur les fléchisseurs du rachis. Le passage en position verticale ne peut se faire qu'en penchant le bassin en avant. On note aussi un décollement bilatéral marqué des omoplates.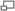
Décrite en 1884 par Louis Landouzy et Jules Dejerine. Atteinte musculaire débutant généralement par la face, progressant à la ceinture scapulaire (muscles maintenant l'omoplate), ensuite aux bras, puis évoluant en touchant les muscles abdominaux, les membres inférieurs et pouvant affecter toute la musculature.
L'atteinte est au début marquée par son asymétrie.
L'âge de début est bien souvent dans l'adolescence, cependant elle peut se déclarer à tout âge, de la première enfance jusqu'à passé 60 ans. Son évolution est généralement lente ou par paliers. Maladie familiale à transmission autosomique dominante. Cas sporadiques dans 30% des cas (sans antécédent familial)
Symptômes additionnels
- Surdité partielle (à totale) dans les sons aigus [1].
- Problème de vascularisation de la rétine.
- problèmes de déglutition[2].
- Atrophie de la langue,
- Atteinte cardiaque[3].
Autres constatations cliniques
- Épisodes inflammatoires, douleurs, biopsies ou EMG montrant des éléments neurogènes.
- Variabilité d'expression de la maladie due aux nombre de répétitions de D4Z4 en 4q35 envisagée
Facteurs aggravants
Réponse immunitaire possible(vaccination ou attaque virale : rage), variation hormonale (adolescence, grossesse, ménopause)[4].
Génétique
-le diagnostic est possible pour 95 % des familles mais il est long et coûteux
Localisation génétique
FSHD1A : L'anomalie était en bout du chromosome 4 sur le bras long en 4q35 dans une région subissant un taux assez fort de réarrangements, c'est cette position en extrémité qui fait que cela a été si long et difficile. Découverte par C. Wijmenga en 1989 (Lancet).
FSHD1B : Des familles FSH ne sont pas liées au chromosome 4, une localisation est supputée sur le chromosome 15 par l'étude de 2 grandes familles (Mark Spers USA).
La fin du chromosome 10 comporte la même structure que le 4, avec un motif de répétition qui ressemble à 98 % à D4Z4, on observe des échanges possibles entre ces parties terminales des chromosomes.
Défaut génétique
En 1992, il est mis en évidence un défaut génétique en relation avec la maladie (faible nombre d'un motif de répétition nommé D4Z4), pas de gène affecté !
L'idée simple : un gène défectueux = une maladie génétique. Là, le problème est autre, on constate un défaut qui coïncide avec la maladie : au bout du chromosome 4, on observe le télomère puis une séquence de 3,3 kb qui est répétée de nombreuses fois, cette séquence est nommée D4Z4. Les patients FSH ont moins de 11 motifs, les personnes saines de 15 à plus de 100.
Premier lien avec la description clinique : il est montré une corrélation entre le faible nombre de motifs de répétition D4Z4 et la sévérité de la maladie ; en résumé moins on a de motifs de répétition plus grand est le risque d'avoir une forme sévère.
Mécanisme génétique
En 2002, 2 publications (Cell, Rossella Tupler et Nature génétics S V D Maarel) : confirment qu'il s'agit d'un problème de répression transcriptionnelle qui ne se fait sur les gènes situés en 4q35 et en amont. (hypothèse supposée depuis 1995). Ces deux publications montrent des éléments différents qui agissent sur la répression transcriptionnelle, l'une avec un ensemble protéique qui se fixe sur le motif de répétition D4Z4 et dont l'effet cumulatif nécessite plusieurs motifs de répétition (au moins une dizaine) pour que la maladie ne s'exprime pas, l'autre qu'il existe deux allèles distinctes 4qA et 4qB et qu'une comporte une séquence effectuant indépendamment la répression transcriptionnelle (du premier mécanisme démontré) ou encore sur l'autre allèle un mécanisme plus subtil.
D'autres facteurs peuvent aussi intervenir : comme le rôle du télomère, nous savons que le télomère est impliqué dans le vieillissement, et agir aussi sur la répression transcriptionnelle.
Quels gènes sont concernés ?
Les gènes candidats FSHD : - Dans D4Z4, un gène Dux4 est potentiellement exprimé. - FRG2 situé à proximité de D4Z4 serait surexprimé dans la FSHD mais ce gène ne semble pas pour autant responsable puisque plusieurs familles FSH qui ont des grandes délétions proximales ont aussi perdu ce gène et développe une FSH tout à fait classiquement. - Dux4C et TUBB4Q, sans avoir prouvé leur sur-expression sont aussi éliminés dans le lien causal par ces familles qui présentent de grandes délétions. - FRG1, rôle dans l’épissage - ALP son rôle avait été exclu - ANT1, adénosine nucléotide translocator , protéine située dans la membrane de la mitochondrie permet la conversion de l’ADP en ATP et lie ainsi la production d’ATP à l’intérieur de la mitochondrie et la consommation d’ATP à l’extérieur de la mitochondrie. Cette protéine joue également un rôle dans le pore de transition de perméabilité de la mitochondrie impliqué dans l’apoptose.
Les autres gènes du chromosome 4, situés en amont sont aussi susceptibles de participer au développement de la pathologie, néanmoins on suppose que des interactions directes peuvent exister avec d’autres chromosomes.
Les gènes extrêmes sont fortement surexprimés et plus on remonte vers le centre, ce dérèglement s'atténue. La pathologie est la conséquence de cette surexpression primitive de ces gènes, qui entraîne ensuite tout un système de compensations se manifestant par des expressions ou sous expressions génétiques secondaires. Déjà, des gènes sont supposés avoir un rôle clef et déterminants dont ANT1 qui régit l'apport énergétique à la cellule mais aussi qui commande l'action des caspases dans l'apoptose (suicide cellulaire). Ce gène, surexprimé, conduit à la mort cellulaire ; mais sous exprimé il devient un facteur cancérigène car des cellules déviantes ne peuvent plus se suicider et prolifèrent en donnant lieu à des cancers. L'étude de moyens de contrôle sur l'expression de ce gène ANT1 est donc particulièrement intéressante.
Histologie
Protéomique
Sarcolemmal reorganization in facioscapulohumeral muscular dystrophy. Ann Neurol. 2006 Feb;59(2):289-97.
- Observations histologiques qui enfin sembleraient montrer quelque chose de spécifique à la FSH. De plus en microscopie électronique, l’écart entre les myofibriles et le sarcolemme est de 5 fois la normale.
- RAGE-NF-kappaB pathway activation in response to oxidative stress in facioscapulohumeral muscular dystrophy. Acta Neurol Scand. 2007 Feb;115(2):115-21.
La protéine ANT 1 est située dans la membrane mitochondriale et permet le passage du flux d’ADP/ATP. Sa sur-expression est susceptible d’accélérer le fonctionnement mitochondrial et de produire davantage d’espèces oxydantes (ROS). On constate un stress oxydatif comme un des premiers événements de la pathologie.
Modèle animal
Aucun modèle animal pour l'instant.
- Le modèle murin FRG1 ne répond que très partiellement à la problématique. Il n'est donc pas validé.
- Le modèle murin PITX1 est dans le même cas "Characterization of a tet-repressible muscle-specific Pitx1 transgenic mouse model as an animal model of FSHD - Yi-Wen Chen"
La création et l’étude de modèles animaux sur-exprimant ces gènes individuellement fournit/fournira la compréhension du rôle de ces gènes, des pistes pour les moduler.
Essais en cours
- Albutérol/Salbutamol (plusieurs études, dont France, USA, NL)
- Prednisone : ouvert USA Tawil 1997 8 patients sur 12 semaines
- Créatine : randomisé, double aveugle, cross over 12 patients sur 8 semaines Walker, 2000
- Créatine (GB Liverpool, Dr Kemp, 2006)
- Dilthiazem (USA, Dr Kissel, 2006)
- Supplémentation en acide folique et méthionine (NL, 2006)
- Inhibiteur de la myostatine M029 USA (en cours et concerne aussi d’autres pathologies)
- Effets d'une supplémentation en antioxydants chez des patients FSHD (en préparation fin 2007), Pr. Mercier - Montpellier
- Autogreffe cellulaire, injections de myoblastes ( En cours, C.Desnuelle, France)
Dans la FSH, les cellules ne se multiplient pas correctement, l'aspect de la culture ressemble à des cellules musculaires normales auxquelles on rajoute un stress oxydatif chimique. Ce qui indique un stress oxydatif endogène, c'est-à-dire un stress oxydatif provoqué par la cellule elle-même. Les cellules poussent vite au départ, puis cela se ralentit très fortement comme si on avait épuisé le potentiel de réplication : on parle de sénescence réplicative. Le dosage de certains ordres biochimiques (myo D, interféron béta ???) laisse supposer une activation précoce du programme myogénique.
Aucun essai thérapeutique n’a encore eu de résultat tangible.
Références
- ↑ Cochlear function in facioscapulohumeral muscular dystrophy. Otol Neurotol. 2007 Jan;28(1):7-10
- ↑ Dysphagia in facioscapulohumeral muscular dystrophy. Neurology. 2006 Jun 27;66(12):1926-8
- ↑ Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur Neurol. 2006;56(1):1-5. Epub 2006 Jun 27
- ↑ Pregnancy and birth outcomes in women with facioscapulohumeral muscular dystrophy. Neurology. 2006 Nov 28;67(10):1887-9
Liens externes
- Amis FSH Europe ::.. - Plus d'info
 Europe - Plus d'infos Site Européen
Europe - Plus d'infos Site Européen- Myopathie facio-scapulo-humérale (FSH) AFM
 Portail de la médecine
Portail de la médecine
Catégories : Maladie neuro-musculaire héréditaire | Myopathie | Maladie génétique
Wikimedia Foundation. 2010.