- Brugada
-
Syndrome de Brugada
Syndrome de Brugada
Classification et ressources externes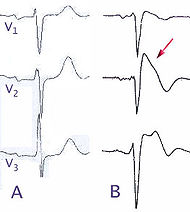
A) ECG normal, dérivation précordial V1 à V3, (B) modification lors d'un syndrome de Brugada (type B) CIM-10 I42.8 CIM-9 746.89 OMIM 601144 DiseasesDB 31999 eMedicine med/3736 MeSH D053840 Le syndrome de Brugada est une maladie génétique rare caractérisée par un sus-décalage du segment ST au niveau des dérivations précordiales droites V1, V2 et V3, et un aspect de bloc de branche droit à l'électrocardiogramme associés à un risque élevé d'arythmie ventriculaire pouvant entrainer syncope et mort subite, sur un cœur structurellement sain. La transmission se fait sur un mode autosomique dominant et la pénétrance est variable. Des mutations génétiques entrainent des anomalies au niveau des canaux ioniques. L'âge moyen du premier épisode clinique est de 40 ans, avec une forte prédominance masculine. La prévalence estimée est d'environ 1/1000 dans les pays asiatiques, probablement plus faible ailleurs. Le pronostic est grave chez les patients présentant des symptômes et la mort subite peut être prévenue par la pose d'un défibrillateur automatique. Ce syndrome a été décrit pour la première fois en 1992 par les frères Pedro et Josep Brugada.
Sommaire
Historique
Le syndrome a été décrit pour la première fois par les frères Brugada[1].
Étiologie
Le syndrome de Brugada est une canalopathie sodique par mutation du gène SCN5A 600163 (en) situé sur le chromosome 3[2]. La mutation sur le gène de l'enzyme glycerol-3 phosphate dehydrogenase-1 like (GPD1-L) provoque une maladie semblable[3].
Il s'agit d'une maladie de transmission autosomique dominante avec une pénétrance faible (peu de porteurs de la mutation présentent les signes de la maladie).
La mutation du gène SCN5A n'est pas la seule cause de la maladie, en effet on peut être porteur d'un syndrome de Brugada (symptomatique ou non) sans avoir de mutation sur le gène SCN5A. Il est possible de mettre en évidence une mutation génétique du SCN5A sur seulement 10 à 25% des patients atteints d'un syndrome de Brugada.Pour les autres patients atteints sans avoir de mutation du SCN5A,la maladie est bien là, mais la mutation d'autres gènes non encore identifiés sont certainement en cause. Des recherches génétiques sont en cours et notamment par le Docteur Ramon Brugada à Montréal.
Épidémiologie
La fréquence est estimée à 1 sur 1000 dans les populations asiatiques où le syndrome de mort brutale au cours du sommeil est fréquent.
L'âge moyen du diagnostic ou de la mort subite est de 40 ans plus ou moins 22 ans, avec des extrêmes allant entre deux jours de vie jusqu'à plus de 80 ans.
Il semble plus fréquent chez les hommes, ces derniers ayant des formes plus graves[4].
Description
Le diagnostic de syndrome de Brugada repose sur l'association d'anomalies à l'ECG (sus-décalage du segment ST), et d'événements de type syncope ou mort subite. Dans un nombre de cas croissant, les cardiologues sont amenés à ce prononcer sur la réalité de ce diagnostic chez des patients asymptomatiques présentant uniquement un aspect anormal à l'ECG (sus-décalage du segment ST).
Diagnostic
Les éléments intervenant dans le diagnostic sont :
- Antécédents familiaux de Syndrome de Brugada, de mort subite ;
- Antécédents personnels de mort subite ressuscitée ;
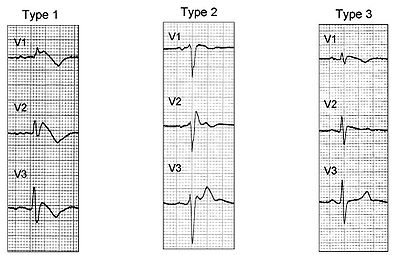
- A l'électrocardiogramme : aspect de bloc de branche droit associé à un sus-décalage du segment ST et des anomalies de l'onde T dans les dérivations précordiales droites (V1 à V3). On décrit 3 types d'anomalies ECG:
- Le type 1: sus-décalage du segment ST en forme de dome.
- Le type 2 : sus-décalage du segment ST en forme de selle de cheval.
- Le type 3 : même figure sur l'électrocardiogramme que le type 2 mais en aplatie.
- Positivité du test à l'Ajmaline ou à la Flécaïne ;
- Identification de la mutation du gène codant SCN5A.
- Électrocardiogramme
 ECG d'un syndrome de Brugada.
ECG d'un syndrome de Brugada.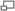
Prise en charge
- Variable, elle dépend du type d'anomalie ECG, du résultat du test pharmacologique, des antécédents familiaux de mort subite, des symptômes et troubles du rythme présentés par le patient.
- En pratique, hormis le cas d'un syndrome de Brugada complet et typique avec antécédent personnel de mort subite et absence de doute diagnostic, indiquant la plupart du temps la pose d'un défibrillateur automatique implantable (DAI), les autres cas (notamment anomalie isolée de l'électrocardiogramme) présentent un risque variable qui est évalué en fonction des examens suscités.
- Il existe actuellement un débat scientifique qui n'est toujours pas tranché sur la conduite à tenir en cas d'aspect électrocardiographique spontané de type 1 chez un patient asymptomatique. Une exploration électrophysiologique est actuellement recommandée avec protocole de stimulation ventriculaire droite programmée afin de rechercher un trouble du rythme ventriculaire déclenché.
- La "sanction" thérapeutique varie, en fonction de la balance entre bénéfices et risques, d'une absence de recommandation particulière (sujet dont on estime que le risque personnel est celui de la population générale saine), jusqu'à la mise en place d'un défibrillateur automatique implantable qui assurera un choc électrique (défibrillation) en cas de survenue de troubles du rythme ventriculaire.
- Il n'existe pas actuellement de traitement médicamenteux prévenant les troubles du rythme ventriculaire dans le syndrome de Brugada.
Conseil génétique
Les données actuelles sur le mécanisme de cette maladie étant en faveur d'un grand nombre de cas de mutations sur les récepteurs de canaux notamment sodiques au sein des cellules myocardiques, il est parfois possible de tenter la recherche de mutation familiales lorsqu'existent des arguments pour de multiples cas familiaux. L'identification de ces mutations, qui n'est pas toujours possible, permet de rechercher si des patients apparentés (parents, enfants) sont porteurs de la mutation, et donc possiblement à risque. Le conseil génétique est utile pour appréhender au cas par cas le risque de transmission à la naissance d'un enfant.
Mode de transmission
Transmission autosomique dominante
Sources
- ↑ Brugada P, Brugada J, Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report, J Am Coll Cardiol, 1992;20:1391-1396
- ↑ Chen Q, Kirsch GE, Zhang D et als. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation, Nature, 1998;392:293–296.
- ↑ London B, Michalec M, Mehdi H et als. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias, Circulation, 2007;116:2260–2268
- ↑ Benito B, Sarkozy A, Mont L et Als. Gender Differences in Clinical Manifestations of Brugada Syndrome, J Am Coll Cardiol, 2008;52:1567-1573
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:601144 [1]
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 [2] Les auteurs de cet article sont les docteurs Brugada.
 Portail de la médecine
Portail de la médecine
Catégories : Canalopathie | Maladie cardio-vasculaire | Trouble du rythme cardiaque | Maladie génétique

Wikimedia Foundation. 2010.