- Rectification (chimie)
-
Distillation fractionnée
 Matériel de laboratoire classique assemblé pour une distillation fractionnée.
Matériel de laboratoire classique assemblé pour une distillation fractionnée.
La distillation fractionnée, aussi appelée rectification, est un procédé de séparation. Son but est de séparer les différents constituants d’un mélange de liquides miscibles, possédant des températures d’ébullition différentes. Pour cela, elle exploite le même principe que la distillation classique mais se distingue par l'utilisation d'une colonne de séparation, qui permet une meilleure discrimination des constituants du mélange.
Sommaire
Matériel requis
- Un ballon rond, contenant le mélange de liquides à séparer
- Un montage à distillation fractionnée: Colonne à fractionnement comme une colonne Vigreux (par exemple avec anneaux de Raschig, un tube de jonction, un adaptateur pour thermomètre, un thermomètre, un réfrigérant, un tube d'écoulement
- Un ballon, une fiole jaugée ou un cylindre gradué pour récupérer le distillat
Mode opératoire
Cas idéal
La solution liquide est chauffée lentement jusqu'à ébullition. Cette ébullition correspond à la vaporisation du composé le plus volatil. Comme lors d'une distillation classique, les vapeurs sont condensées pour obtenir un produit A pur, collecté dans un premier récipient. La solution liquide est alors exempte du produit A. On change le récipient de récupération et on augmente la température du mélange liquide afin de recueillir chaque constituant séparément (B, C, ...). On repère plusieurs paliers de température correspondant à la vaporisation des différents constituants du mélange initial. On a donc autant de paliers de température que de constituants.
Cas réel
Souvent, les liquides à séparer ont soit des températures d'ébullition très proches, soit de grandes affinités. C'est le cas de l'eau et de l'éthanol qui forment un azéotrope, c'est à dire un mélange possédant sa propre température d'ébullition. Lors du chauffage, le composé arrivant en tête de de colonne vers 78,1 °C, est un mélange contenant environ 95% d'éthanol. C'est dans ce genre de situations que l'on a recours à la distillation fractionnée pour "casser" l'azéotrope et séparer les deux constituants.
Cependant lorsque le mélange azéotropique à séparer est positif (température d'ébullition de l'azéotrope inférieure aux composés purs) ou négatif (température d'ébullition supérieure), il n'est possible de purifier qu'un seul des deux composés.
Distillation fractionnée
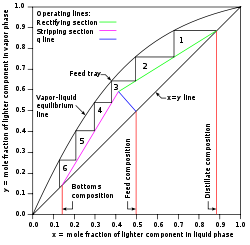 Diagramme de McCabe-Thiele utilisé pour calculer le nombre de plateau théorique suffisant à la séparation d'un mélange de deux constituants. Cliquez sur l'image pour lire.
Diagramme de McCabe-Thiele utilisé pour calculer le nombre de plateau théorique suffisant à la séparation d'un mélange de deux constituants. Cliquez sur l'image pour lire.Lorsque les vapeurs montent dans la colonne de séparation, elles se refroidissent et se condensent sur la surface interne de la colonne (les aiguilles de la colonne de Vigreux ou les anneaux de Raschig d'une colonne garnie). Ce liquide est ensuite chauffé progressivement par les autres vapeurs montantes jusqu'à être vaporisé à nouveau. Toutefois, la composition de ces nouvelles vapeurs n'est pas la même que celle des vapeurs initiales (voir la loi de Raoult): elles sont plus concentrées en composant le plus volatil.
Chaque cycle de vaporisation-condensation se produisant au sein de la colonne de séparation (appelé un plateau théorique) conduit à une augmentation de la concentration en composé le plus volatil. On peut donc caractériser la colonne par son nombre de plateaux théoriques: plus celui-ci est élevé, plus la colonne sera capable de fractionner le mélange avec finesse. Cette méthode a été découverte par McCabe et Thiele en 1925.
On augmente le nombre de plateaux théoriques en allongeant la colonne ou en modifiant sa surface interne grâce à différents types de garnissages.
Remarque: Il est aussi possible de travailler à pression réduite pour abaisser le point d'ébullition des composés. On parle alors de distillation sous vide.
Articles connexes
 Portail de la chimie
Portail de la chimie
Catégorie : Procédé chimique


Wikimedia Foundation. 2010.