- Méthode GF de Wilson
-
Méthode GF
La méthode GF de Wilson, parfois appelée méthode FG, est une méthode de mécanique classique d'obtention de certaines coordonnées internes pour une molécule semi-rigide vibrante, les coordonnées normales Qk. Les coordonnées normales décomposent les mouvements classiques de vibrations de la molécule et permettent ainsi d'obtenir des amplitudes vibrationnelles des atomes en fonction du temps. On postule, dans la méthode GF de Wilson, que l'énergie cinétique moléculaire est due seulement aux vibrations harmoniques des atomes, c'est-à-dire que l'énergie globale de rotation et translation est ignorée. Les coordonnées normales apparaissent aussi dans la description quantique des mouvements de vibration de la molécule et dans le couplage de Coriolis entre rotations et vibrations.
Il en découle qu'après application des conditions d'Eckart que la matrice G-1 donne l'énergie cinétique en termes de coordonnées internes linéaires arbitraires, alors que F représente l'énergie potentielle (harmonique) en fonction de ces coordonnées. La méthode GF donne la transformation linéaire des coordonnées internes générales à l'ensemble spécial de coordonnées normales.
Sommaire
La méthode GF
Une molécule non-linéaire de N atomes possède 3N-6 degrés de liberté internes, car localiser une molécule dans un espace tridimensionnel nécessite trois degrés de liberté et son orientation spatiale nécessite trois autres degrés de liberté. Ces degrés de liberté doivent être soustraits des 3N degrés de liberté d'un système de N particules.
Les atomes d'une molécule sont « liés » par une surface d'énergie potentielle (SEP) (ou un champ de force dans une approche classique)[1] qui est une fonction de 3N-6 coordonnées.
Les degrés internes de liberté q1, ..., q3N-6 décrivant la SEP de façon optimale sont parfois non-linéaires, comme les coordonnées de valence telles que les angles de liaison, de torsion ou les étirements de liaisons. Il est possible d'écrire l'opérateur d'énergie cinétique quantique pour de telles coordonnées curvilignes, mais il est difficile de formuler une théorie générale applicable à toute molécule. C'est pourquoi E.B. Wilson[2] linéarisa les coordonnées internes en postulant des déplacements minimes. La version linéarisée de la coordonnée interne qt est notée St.
La SEP V peut être développée en série de Taylor au voisinage de son minimum en termes de St. Le troisième terme (le hessien de V), évalué au minimum, est une matrice 'F dérivée de la force. Dans l'approximation harmonique, la série de Taylor s'achève avec ce terme. Le deuxième terme, contenant les dérivées premières, est nul car évalué au minimum de V. Le premier terme peut être inclus dans le zéro d'énergie. D'où :
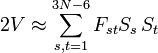 .
.
L'énergie cinétique de vibration classique a la forme :
où gst est un élément du tenseur métrique des coordonnées internes (curvilignes). Les points indiquent les dérivées temporelles. L'évaluation du tenseur métrique g au minimum q0 de V donne la matrice définie positivement et symétrique G = g(q0)-1. On peut alors résoudre les deux problèmes matriciels suivants simultanément :
car ils sont équivalents au problème aux valeurs propres généralisé :
où
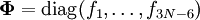 et
et  est la matrice unitaire. La matrice L-1 contient les coordonnées normales Qk dans ses lignes :
est la matrice unitaire. La matrice L-1 contient les coordonnées normales Qk dans ses lignes :En raison de la forme du problème aux valeurs propres généralisé, la méthode est appelée méthode GF, appellation à laquelle on accole parfois le nom de son inventeur : la méthode GF de Wilson.
On introduit les vecteurs
satisfaisant la relation :
En introduisant les résultats de l'équation aux valeurs propres, l'énergie de la molécule E = T + V (dans l'approximation harmonique) devient :
Le lagrangien L = T - V est :
Les équations de Lagrange sont identiques aux équations de Newton :
pour un ensemble d'oscillateurs harmoniques non couplés. Ces équations différentielles ordinaires du second ordre sont facilement solvables, et donnent Qt en fonction du temps (voir l'article sur l'oscillateur harmonique).
Coordonnées normales en termes de coordonnées cartésiennes de déplacement
Les coordonnées normales sont parfois exprimées comme des combinaisons linéaires des coordonnées cartésiennes de déplacement. Soit RA le vecteur position du noyau A et RA0 la position à l'équilibre correspondante.
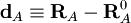 est par définition la coordonnée cartésienne de déplacement du noyau A. La linéarisation de Wilson des coordonnées curvilignes internes qt exprime la coordonnée St en termes de coordonnées de déplacement :
est par définition la coordonnée cartésienne de déplacement du noyau A. La linéarisation de Wilson des coordonnées curvilignes internes qt exprime la coordonnée St en termes de coordonnées de déplacement :où sAt est le vecteur s de Wilson. Si l'on introduit
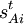 dans la matrice 3N-6 x 3N B, cette équation devient en notation matricielle :
dans la matrice 3N-6 x 3N B, cette équation devient en notation matricielle :La forme réelle des éléments de matrice de B peut être relativement compliquée. Pour un angle de torsion en particulier, impliquant 4 atomes, on a besoin d'une algèbre vectorielle fastidieuse afin d'en déduire les valeurs correspondantes des
. On pourra, pour plus d'informations sur cette méthode connue sous le nom de méthode des vecteurs s de Wilson se reporter à l'ouvrage de Wilson et collaborateurs, ou à l'article vibration moléculaire. À ce stade,Sous forme de somme :
D est ici une matrice 3N-6 x 3N donnée par la linéarisation des coordonnées internes q (procédé algébrique) et la solution des équations GF de Wilson (procédé numérique).
Relations aux conditions d'Eckart
Article principal : Conditions d'Eckart.A partir de l'invariance des coordonnées internes St par rapport à toute rotation globale et translation de la molécule, on en déduit que les coordonnées linéarisées stA présentent les mêmes propriétés.On peut montrer que cela implique que les six conditions suivantes sont satisfaites par les coordonnées internes :
Ces conditions découlent des conditions d'Eckart qui sont valables pour les vecteurs déplacement :
Notes
- ↑ De manière générale, la SEP décrit les variations d'énergie induites par les différentes interactions entre atomes. Les positions atomiques y sont localisées dans des « vallées » où ces énergies sont minimales, et d'où il peut être difficile de s'extraire sauf apport extérieur d'énergie (excitations électromagnétiques ou thermiques, etc.).
- ↑ E. B. Wilson, Jr. Some Mathematical Methods for the Study of Molecular Vibrations, J. Chem. Phys. vol. 9, pp. 76-84 (1941)
Références
- (en) E. B. Wilson, J. C. Decius, et P. C. Cross, Molecular Vibrations, McGraw-Hill, New York, 1955 (réimprimé par Dover, 1980).
- (en) D. Papoušek et M. R. Aliev, Molecular Vibrational-Rotational Spectra Elsevier, Amsterdam, 1982.
- (en) S. Califano, Vibrational States, Wiley, London, 1976.
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « GF method ».
 Portail de la chimie
Portail de la chimie Portail de la physique
Portail de la physique
Catégories : Spectroscopie | Chimie quantique
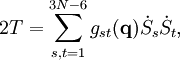
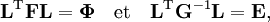
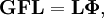
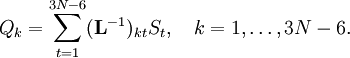
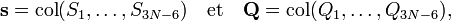
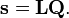
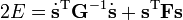
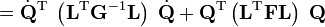
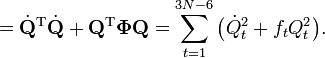
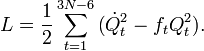
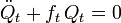
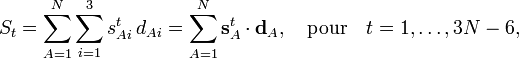
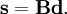
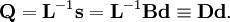
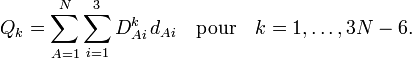
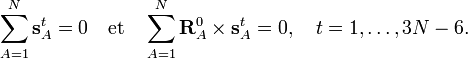
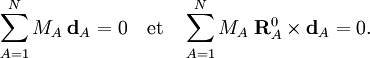
Wikimedia Foundation. 2010.