- Épidermolyse bulleuse
-
Épidermolyse bulleuse épidermolytique Référence MIM 131760-131800
131900-131960
601001Transmission Dominante principalement Chromosome 17q12-q21-12q13 Gène KRT14-KRT5 Empreinte parentale Non Mutation Ponctuelle Mutation de novo Possible Incidence 1 sur 50000 naissances Maladie génétiquement liée Aucune Diagnostic prénatal Possible Liste des maladies génétiques à gène identifié modifier 
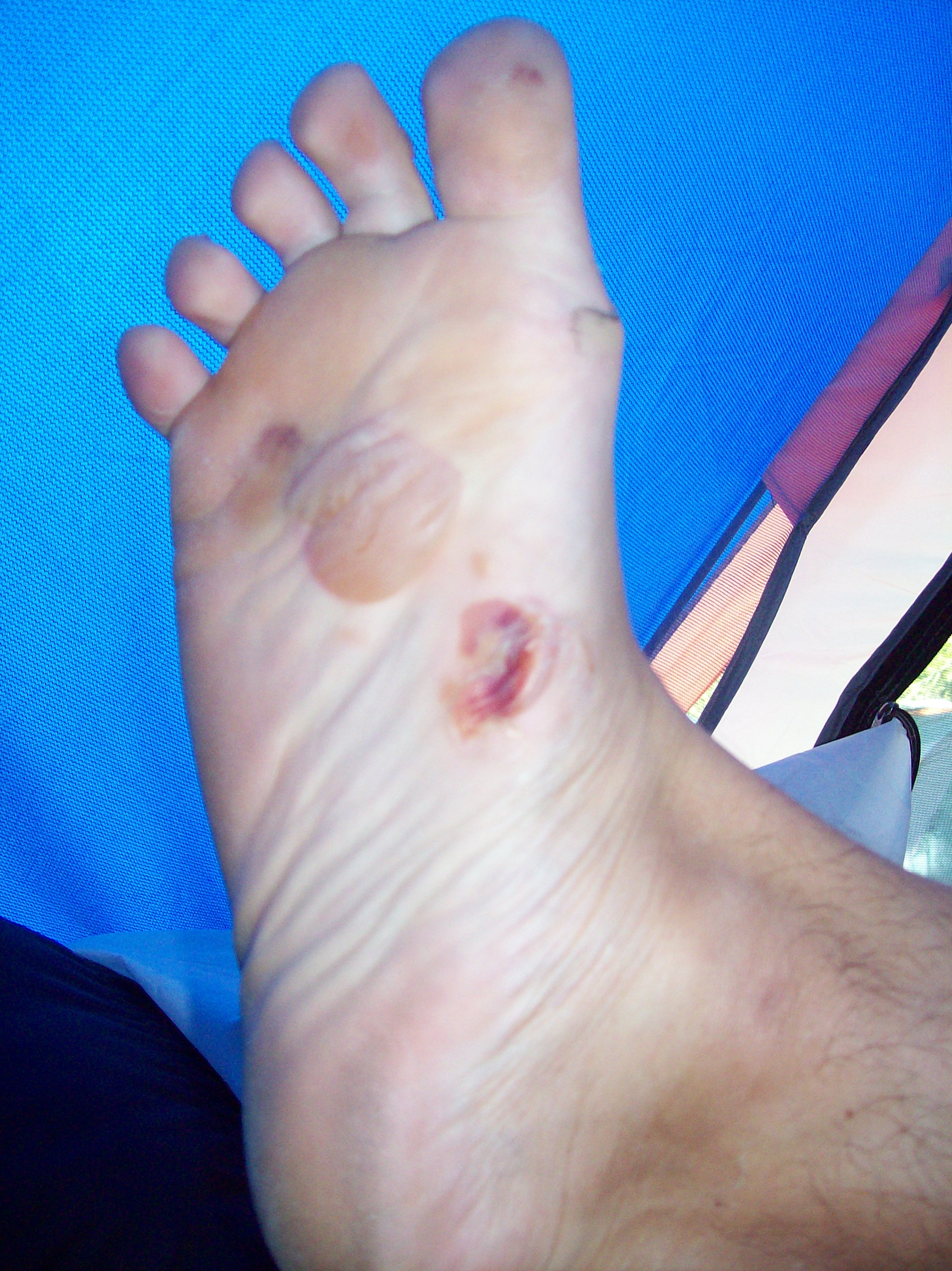
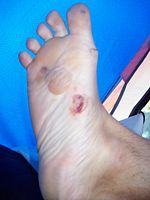 Ampoules formées sur le pied d'une personne souffrant d'épidermolyse bulleuse épidermolytique de type Weber-Cockayne.
Ampoules formées sur le pied d'une personne souffrant d'épidermolyse bulleuse épidermolytique de type Weber-Cockayne.
L' épidermolyse bulleuse épidermolytique est une maladie génétique se caractérisant par une fragilité de la peau et se traduisant par l'apparition de bulles ou de vésicules épidermiques pour des traumatismes minimes de la peau.
Sommaire
Types cliniques
Il existe plus de 20 types différents d'EB dont quatre types cliniques sont connus:
- Épidermolyse bulleuse épidermolytique type Weber-Cockayne avec des bulles uniquement au niveau des pieds et des mains. Les manifestations commencent rarement à la naissance. Les premiers signes apparaissent généralement vers 18 mois et parfois dans l'adolescence.
- Épidermolyse bulleuse épidermolytique type Koebner où les premières bulles apparaissent dans les premiers mois de vie mais peuvent être aussi présentes à la naissance. Les zones du corps atteintes sont nombreuses.
- Épidermolyse bulleuse épidermolytique avec pigmentation mouchetée où des tâches brunes apparaissent alternant avec des zones hypopigmentées surtout au niveau du tronc ou des mains. Ces troubles de la pigmentation disparaissent à l'âge adulte.
- Épidermolyse bulleuse épidermolytique type Dowling-Meara qui peut être mortelle. Les bulles sont évidentes à la naissance.
Une autre classification la divise en trois groupes principaux selon ses manifestations: l'EB simple, l'EB dystrophique et l'EB fonctionnelle
Diagnostic de la maladie
Le diagnostic du type Weber-Cockayne est presque toujours fait par la clinique. Les autres formes nécessitent la biopsie d'une bulle récente, l'étude au microscope électronique ou avec l'aide anticorps fluorescents met en évidence les lésions de la maladie. Le diagnostic génétique est possible mais rarement utilisé en pratique clinique. Il est surtout utilisé pour le conseil génétique.
Évolution de la maladie
La sévérité des manifestations varie grandement à la fois dans une même famille et entre les individus, soit d'une forme peu sévère à des formes mutilantes, voire mortelles. Un «accident» génétique provoque une défaillance du système d'adhésion des cellules dermiques et des différentes couches de l'épiderme. La peau est alors extrêmement fragile et se décolle au moindre frottement. Des multiples petites vésicules agglutinées sont typiques de la maladie et la survenue de bulle hémorragique est habituelle dans cette maladie. La douleur existe en permanence mais elle varie beaucoup en fonction du type d'EB. Une amélioration peut survenir tardivement dans l'enfance. La chaleur améliore la maladie chez certains individus.
Une hyperkératose des mains et des pieds survient dans l'enfance et peut devenir une source de plaintes majeures chez les personnes atteintes dans la vie adulte. Des troubles de la croissance des ongles et un millium cutanée sont habituels.
Des troubles de l'alimentation traduisent une atteinte des muqueuses digestives.
Soins
La prise en charge de cette pathologie comprend la protection de la peau contre les traumatismes et la prévention des infections secondaires. L'activité des enfants devra diminuer les risques de traumatisme et les nouvelles bulles devront être percées. L'habillement se fera en trois couches : Une non adhérente au contact de la peau, une deuxième protection rembourrée et la troisième élastique. L'utilisation du chlorure d' aluminium et du cyproheptadine peut réduire la formation de bulles chez certains individus. L'utilisation de kératolytique et d'adoucissant de la peau permet de lutter contre l'hyperkératose.
Source
- (en) Ellen G Pfendner, Anne W Lucky, Epidermolysis Bullosa Simplex In GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005 [1]
Wikimedia Foundation. 2010.