- Théorie de la perturbation de møller-plesset
-
Théorie de la perturbation de Møller-Plesset
Méthodes numériques pour le calcul de la structure électronique
Hartree-Fock Théorie de la perturbation de Møller-Plesset Interaction de configuration Méthode du cluster couplé Champ multi-configurationnel auto-cohérent Théorie de la fonctionnelle de la densité La théorie de la perturbation de Møller-Plesset (MP) est une des nombreuses méthodes post-Hartree-Fock ab initio en chimie quantique appliquée dans le cadre de la chimie numérique. Elle améliore la méthode Hartree-Fock en y apportant les effets de corrélation électronique au moyen de la théorie de la perturbation de Rayleigh-Schrödinger (RS-PT) au deuxième (MP2), troisième (MP3) ou quatrième (MP4) ordre habituellement. L'idée principale de cette méthode a été publiée dès 1934[1].
Sommaire
Théorie de la perturbation de Rayleigh-Schrödinger
La théorie MP est une application particulière de la théorie de la perturbation de Rayleigh-Schrödinger (en anglais Rayleigh-Schrödinger perturbation theory, RS-PT). Dans la RS-PT, on considère un opérateur hamiltonien non perturbé
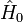 auquel est ajouté une petite perturbation (parfois externe)
auquel est ajouté une petite perturbation (parfois externe) 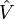 :
: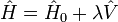 ,
,
où λ est un paramètre arbitraire réel. Dans la théorie MP, la fonction d'onde d'ordre 0 est une fonction propre exacte de l'opérateur de Fock, qui sert alors d'opérateur non perturbé. La perturbation est le potentiel de corrélation.
Dans la RS-PT, la fonction d'onde perturbée et l'énergie perturbée sont exprimées sous forme de séries entières de λ :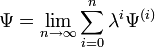 ,
,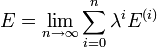 .
.
L'inroduction de ces séries dans l'équation de Schrödinger dépendante du temps donne une nouvelle équation :
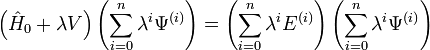 .
.
L'égalisation des facteurs des λk dans cette équation donne l'équation de la perturbation d'ordre k, où k=0,1,2, ..., n.
Perturbation de Møller-Plesset
Formulation initiale
Les corrections d'énergie MP sont obtenues à partir de la théorie de la perturbation de Rayleigh-Schrödinger (RS) avec la perturbation (potentiel de corrélation) :
dans laquelle le déterminant de Slater normalisé Φ0 est la fonction propre la plus basse de l'opérateur de Fock :
Ici, N est le nombre d'électrons de la molécule considérée, H est le hamiltonien électrique habituel, f(1) est l'opérateur de Fock mono-électronique, et εi est l'énergie orbitalaire appartenant à l'orbitale spatiale doublement occupée φi. L'opérateur de Fock décalé
sert d'opérateur non perturbé (ordre zéro).
Le déterminant de Slater Φ0 étant une fonction propre de F, il s'ensuit que :
donc que l'énergie à l'ordre zéro est la valeur attendue de H en fonction de Φ0, 'i.e. l'énergie Hartree-Fock :
Comme l'énergie MP de premier ordre :
est bien sûr nulle, l'énergie de corrélation MP apparait dans le terme de second ordre. Ce résultat est le théorème de Møller-Plesset[1]: « Le potentiel de corrélation ne contribue pas à l'énergie électronique exacte dans le terme de premier ordre ».
Afin d'obtenir la formule MP2 pour une molécule quasiment isolée, la formule de second ordre RS-PT est écrite sur la base de déterminants de Slater doublement excités (les déterminants de Slater mono-excités ne contribuent pas en raison du théorème de Brillouin). Après application des lois de Slater-Condon pour la simplification des éléments de matrices N-électroniques des déterminants de Slater en bra et ket et en intégrant le spin, on obtient :
où phi;i et φj sont les orbitales canoniques occupées et φa et φb sont les orbitales canoniques virtuelles. Les quantités εi, εj, εa, et εb sont les énergies orbitalaires correspondantes. Ainsi, par le terme de second ordre dans le potentiel de corrélation, l'énergie électronique totale est donnée par l'énergie de Hartree-Fock et la correction de second ordre MP : E ≈ EHF + EMP2. La solution de l'équation MP à l'ordre zéro (qui par définition est l'équation de Hartree-Fock) donne l'énergie de Hartree-Fock. La première correction de perturbation au-delà du traitement Hartree-Fock qui ne s'annule pas est le terme d'énergie de second ordre.
Formulation alternative
Des expressions équivalentes sont obtenues par une partition légèrement différente du hamiltonien, qui donne une séparation différente des termes d'énergie sur les contributions d'ordre zéro et un, alors qu'à des ordres plus élevés, les deux séparations donnent des résultats identiques. Cette formulation est largement répandue les chimistes, qui sont maintenant de grands utilisateurs de ces méthodes[2]. Cette différence est due au fait, bien connu en théorie Hartree-Fock, que :
(l'énergie Hartree-Fock n'est pas égale à la somme des énergies des orbitales occupées). Dans la séparation alternative, on définit :
On a de manière évidente, dans cette séparation :
Le théorème de Møller-Plesset n'abonde pas dans le sens où EMP1 ≠ 0. La solution à l'équation MP d'ordre zéro est la somme des énergies orbitalaires. La correction d'ordre zéro plus un donne l'énergie Hartree-Fock. Comme dans la formulation initiale, le premier terme de perturbation qui ne s'annule pas au-delà du traitement Hartree-Fock est l'énergie de second ordre, qui est, rappelons-le, le même dans les deux formulations.
Utilisation des méthodes de perturbation Møller-Plesset
Les calculs Møller-Plesset du second (MP2), du troisième (MP3) et quatrième ordre (MP4) sont les niveaux standard utilisés pour des petits systèmes et sont implémentés dans de nombreux codes de chimie numérique. Des calculs MP de niveau plus élevés, en général seulement du MP5, sont disponibles dans certains codes. Cependant, ils sont rarement utilisés en raison de leurs coûts.
Des études systématiques sur la théorie de la perturbation MP ont montré qu'elle n'est pas forcément convergente à des ordres élevés. Les propriétés de convergence peuvent être lentes, rapides, oscillantes, régulières, hautement erratiques ou simplement inexistantes, selon le système chimique étudié ou la base utilisée. [3] De plus, diverses propriétés moléculaires importantes calculées au niveau MP3 et MP4 ne sont en aucune façon meilleur que leurs équivalents MP2, même pour de petits systèmes[4]. Pour les molécules à couches ouvertes, la théorie MP d'ordre n ne peut être appliquée directement qu'aux fonctions de référence la méthode de Hartree-Fock non restreinte (les états de la méthode Hartree-Fock restreinte ne sont pas des vecteurs propres généraux de l'opérateur de Fock). Cependant, les énergies résultantes « souffrent » d'une sévère contamination de spin, conduisant à des résultats complètement erronés. Une meilleure alternative est d'utiliser une des méthodes similaires à la méthode MP2 basées sur des références Hartree-Fock restreinte pour couche ouverte.
Ces méthodes, Hartree-Fock, Hartree-Fock non-restreinte et Hartree-Fock restreinte utilisent une fonction d'onde à déterminant simple. Les méthodes à champ auto-cohérent multi-configurationnel utilisent plusieurs déterminants et peuvent être utilisées pour l'opérateur non-perturbé, bien qu'il n'y ait pas qu'une seule méthode d'application, comme la théorie de la perturbation spatiale complète (Complete Active Space Perturbation Theory, CASPT2).Voir aussi
Références
- ↑ a et b (en) C. Møller et M.S. Plesset, « Note on an Approximation Treatment for Many-Electron Systems », dans Phys. Rev., vol. 46, 1934, p. 618–622 [texte intégral lien DOI]
Cet article de nombreux problèmes mathématiques mineurs, mais cependant ennuyeux, tel que publié. Se référer un bon manuel de chimie quantique pour une dérivation concise à l'ordre n..
- ↑ Voir toutes les références de la section Lectures complémentaires.
- ↑ Leininger M.L., Allen W.D., Schaefer H.F., Sherrill C.D., « Is Moller-Plesset perturbation theory a convergent ab initio method? », dans J. Chem. Phys., vol. 112, no 21, 2000, p. 9213-9222
- ↑ Trygve Helgaker, Poul Jorgensen et Jeppe Olsen, « Molecular Electronic Structure Theory », Wiley (2000), ISBN 978-0471967552.
Lectures complémentaires
- Christopher J. Cramer, « Essentials of Computational Chemistry » (John Wiley & Sons, Ltd., 2002), p. 207-211, ISBN 0-471-48552-7
- James B. Foresman et Æleen Frisch, « Exploring Chemistry with Electronic Structure Methods », (Gaussian Inc., 1996), p. 267-271, ISBN 0-9636769-4-6
- Andrew R. Leach, « Molecular Modelling » (Longman, 1996), p. 83-85, ISBN 0-582-23933-8
- Ira N. Levine, « Quantum Chemistry » (Prentice Hall, 1991), p. 511-515, ISBN 0-205-12770-3
- Attila Szabo et Neil S. Ostlund, « Modern Quantum Chemistry », (Dover Oublications, Inc, 1996), p. 350-353, ISBN 0-486-69186-1
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « Møller-Plesset perturbation theory ».
 Portail de la chimie
Portail de la chimie Portail de la physique
Portail de la physique
Catégories : Chimie quantique | Chimie numérique
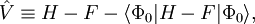
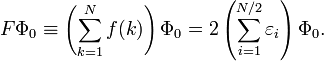
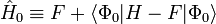
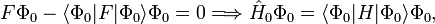
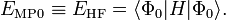
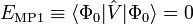
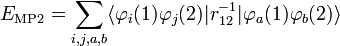
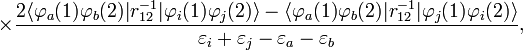
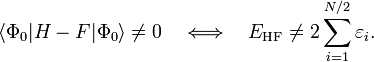
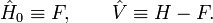
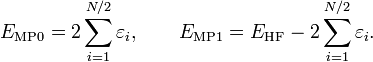
Wikimedia Foundation. 2010.