- Theorie de la cinetique d'oxydation de Wagner
-
Théorie de la cinétique d'oxydation de Wagner
La Théorie de la cinétique d'oxydation de Wagner est une théorie physique destinée à modéliser la vitesse de croissance d'une couche d'oxyde sur un métal lorsque celle-ci est compacte et adhérente, dans le cadre de la corrosion sèche. Elle fut proposée par Carl Wagner en 1933.
Cette théorie prédit une loi parabolique : la masse oxydée, ou la prise de masse Δm, est proportionnelle à la surface réactive S et à la racine carrée du temps t :
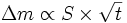 .
.
Ce comportement en racine carrée du temps est également constaté expérimentalement. Intuitivement, on peut concevoir que la croissance est de moins en moins rapide car les espèces chimiques doivent traverser une couche d'oxyde de plus en plus épaisse.
Le calcul fait par Wagner prend en compte la diffusion sous l'effet de l'agitation thermique, les charges des espèces et notamment la force électrostatique qui s'y exerce, ainsi que les équations thermochimiques qui résultent du quasi-équilibre chimique à chaque point de l'oxyde.
Cependant, cette loi fait des hypothèses trop restrictives, et la cinétique calculée à partir de cette théorie ne correspond pas aux résultats expérimentaux (la forme de la courbe correspond bien, mais pas l'échelle). Elle reste malgré tout la loi la plus complète et sert de base à d'autres extrapolations prenant en compte, par exemple, l'effet des joints de grain.
Sommaire
Introduction
Pour traiter le problème de la cinétique d'oxydation à haute température, Wagner[1] a fait les hypothèses suivantes quant à la migration des espèces dans l'oxyde[2],[3],[4] :
- la migration fait intervenir, outre le déplacement par sauts aléatoires (diffusion), l'effet du gradient de potentiel chimique ainsi que l'effet du champ électrique local créé par la répartition des charges ;
- l'oxyde a une composition proche de la stœchiométrie ;
- à tout instant, l'oxyde est localement à l'équilibre chimique ;
- le circuit est ouvert, c'est-à-dire que le courant électrique global est nul et donc que les flux d'espèces chargées sont couplés.
La formation d'oxyde se fait par diffusion d'ions, le but est donc de calculer le flux ionique en fonction des paramètres fondamentaux de l'oxyde (coefficient de diffusion, conductivités, pression partielle d'oxygène).
Prérequis
Les prérequis pour cette étude sont :
- la loi de Fick, loi phénoménologique de la diffusion ;
- la loi de Nernst-Einstein, résultant de l'étude statistique du mouvement d'une particule sous l'effet combiné d'une force et de chocs aléatoires ;
- la loi d'Ohm sous sa forme microscopique ;
- la conduction électrique dans les oxydes cristallins
- la relation thermodynamique de Gibbs-Duhem
Convention de notation et stratégie
On considère que l'oxyde a pour formule :
- MnO2
où « M » est un métal. On peut dans une première approche considérer que c'est un cristal ionique, les ions étant alors :
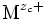 (parfois noté Mzc + ) et O2-.
(parfois noté Mzc + ) et O2-.
La neutralité électrique du cristal impose qu'il y ait autant de charges positives que de charges négatives, soit
- n × zc = 2 × 2 = 4 (E1)
Il s'agit là de la neutralité électrique « constitutive », « stœchiométrique », de l'oxyde. Lors de l'oxydation, il se forme d'autres charges (comme les électrons libres et les trous d'électron), il y a donc une autre équation de la neutralité électrique qui prend en compte les concentrations des différentes espèces.
On considère qu'une « cellule élémentaire » MnO2 se forme par la rencontre de n ions métalliques Mzc + et de deux ions oxygène O2- selon la réaction :
- n Mzc + + 2 O2- → MnO2.
Le but du présent article est donc de présenter un calcul du flux d'ions (nombre d'ions traversant une surface unité perpendiculaire au flux, c'est-à-dire parallèle à la surface libre de l'échantillon, durant un temps unité) :
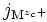 étant le flux de cations métalliques vers l'extérieur,
étant le flux de cations métalliques vers l'extérieur, 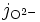 étant le flux d'anions oxygène vers l'intérieur.
étant le flux d'anions oxygène vers l'intérieur.
Un flux de particules i chargées crée un courant électrique. Si chaque particule porte zi charges élémentaires e, alors la densité de courant électrique jel i est reliée au flux de particules ji par la formule :
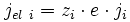 (E2)
(E2)
Dans le cas des ions dans l'oxyde, le flux de cations vers l'extérieur et le flux d'anions vers l'intérieur crée un courant électrique nommé « courant ionique », noté jion, et valant :
Si la réaction de formation est très rapide devant la migration des espèces, alors pour une surface S, la vitesse v de formation de l'oxyde (en mole par seconde) est :
ce qui, d'après l'électroneutralité (E1) devient
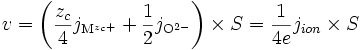 (E3)
(E3)
Nous allons donc chercher à déterminer le courant ionique afin de déterminer la vitesse de croissance de l'oxyde.
Migration sous l'effet d'un champ
Les espèces effectuant des sauts aléatoires, nous considérons le parcours moyen <X> d'une particule (atome, ion, électron libre, trou d'électron) pendant une durée τ. On peut définir la vitesse moyenne v comme étant
Si la particule est soumise à une force F, alors on peut relier la vitesse moyenne à la force et au coefficient de diffusion D par la loi de Nernst-Einstein :
k étant la constante de Boltzmann et T la température absolue. Si l'on prend la comparaison avec le frottement fluide (freinage du parachute), on peut écrire que la vitesse est proportionnelle à la force :
- v = B⋅F
B étant la mobilité (Beweglichkeit) de l'espèce. On a donc la relation d'Einstein :
- D = B⋅k⋅T (E4)
Le flux j, lui, résulte du mouvement sous l'effet de la force, qui tend à faire migrer les espèces et donc à créer un gradient de concentration, et de l'agitation thermique qui tend elle au contraire à uniformiser la concentration (loi de Fick). Comme on se place dans le cas d'un oxyde dont la composition est proche de la stœchiométrie, on suppose également que la variation de concentration est faible. Le flux peut donc s'écrire, par approximation :
- j = v⋅c = c⋅B⋅F (E5)
c étant la concentration.
Potentiel chimique
La force Fc résultant du potentiel chimique μ peut s'écrire, à une dimension :
et donc (E5) devient :
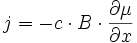 (E6)
(E6)
Champ électrique
Si une particule porte z charges élémentaires e, alors elle subit la force Fe (force électrostatique ou force de Coulomb) :
Le champ électrique E dérive d'un potentiel V, ce qui s'écrit à une dimension :
d'où, d'après (E5), un flux :
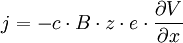 (E7)
(E7)
soit une densité de courant électrique (flux de charges) jel[5] valant d'après (E2)
On peut faire un parallèle avec le loi d'Ohm reliant cette densité de courant électrique jel au gradient de potentiel[6] :
σ étant la conductivité électrique. On a donc :
soit
- σ = c⋅B⋅z²⋅e² (E8)
et donc d'après la relation d'Einstein (E4) :
Lorsqu'il y a plusieurs porteurs de charge i, la conductivité totale σ est la somme des conductivités pour chaque espèce σi :
- σ = ∑i σi
On peut aussi écrire que chaque σi est une portion ti de σ :
- σi = ti⋅σ
avec évidemment
- ∑i = ti = 1
Le coefficient ti est appelé le « nombre de transport » de l'espèce i.
On obtient donc pour une espèce donnée :
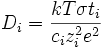 (E9)
(E9)
Équilibre thermodynamique
Dans cette partie, nous considèrerons pour simplifier que la migration, pour le métal ou bien l'oxygène, se fait par une seule espèce (ion interstitiel ou bien lacune, cf. Défaut cristallin et Notation de Kröger et Vink), et nous noterons :
- M pour désigner l'ion métallique en position normale dans l'oxyde MM ;
- Mzc+ pour désigner l'ion mobile dans l'oxyde, c'est-à-dire soit l'ion interstitiel Mizc•, soit la lacune métallique VMzc ' ;
- O pour désigner l'ion oxygène en position normale OO ;
- O2- pour désigner l'ion mobile dans l'oxyde, Oi2 ' ou VO2• ;
- e- pour désigner les électrons libres e' et h+ les trous d'électrons h•.
Notez que lorsqu'une lacune se déplace, cela équivaut globalement au déplacement en sens inverse d'un ion de charge opposée à la charge effective de la lacune.
Si l'on écrit la relation de Gibbs-Duhem, on obtient :
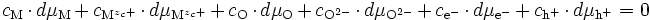 (E10)
(E10)
Écrivons maintenant les équilibres chimiques locaux :
- cations : M ⇌ Mzc+ + zc e- ⇒ dμM = dμMzc+ + zc dμe- (E11) ;
- anions : O + 2e- ⇌ O2- ⇒ dμO + 2 dμe- = dμO2- (E12);
- électrons et trous d'électron : Ø ⇌ e- + h+ ⇒ 0 = dμe- + dμh+ soit dμe- = - dμh+ (E13).
La relation de Gibbs-Duhem (E10) s'écrit donc :
L'électroneutralité s'écrit :
donc
Puisque nous sommes proches de la stœchiométrie, on peut écrire la neutralité électrique du cristal : l'ensemble des cations (en position interstitielle ou en position normale) contrebalance l'ensemble des anions (en position interstitielle ou en position normale) en ce qui concerne les charges :
La relation de Gibbs-Duhem devient donc au final
ou bien encore
et donc
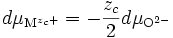 (E14)
(E14)
Courant ionique
Les espèces chargées migrent sous l'effet du gradient de potentiel chimique et électrique, donc pour une espèce i (E6) (E7) :
Ce flux crée un courant électrique dont la densité vaut (E2) (E6) (E7) :
soit d'après (E8):
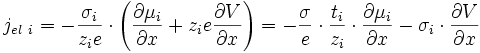 (E15).
(E15).
puisque σi = ti·σ.
Le circuit est ouvert : il n'y a pas de circulation de charges à l'extérieur de l'oxyde, on a donc :
soit
Comme on a
-
∑ σi = σ i
et avec (E13), on obtient :
et d'après (E14) :
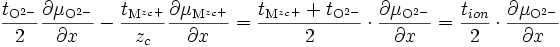 (E16)
(E16)
si l'on note le nombre de transport ionique tion = tMzc+ + tO2- et le nombre de transport électrique tel = te- + th+. On obtient :
et d'après (E12)
comme tion + tel = 1, on obtient
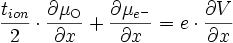 (E17)
(E17)
La quantité qui nous intéresse est le courant ionique, qui s'écrit d'après (E15) :
soit d'après (E16)
en utilisant l'équilibre thermodynamique de l'oxygène (E12) et en remplaçant
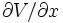 par l'expression trouvée en (E17), on a :
par l'expression trouvée en (E17), on a :soit
(E18)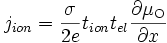
Vitesse de croissance
où v(x) est le nombre de cellules de MnO2 crées en une seconde à la profondeur x par rapport à la surface (toutes les grandeurs sont en effet locales).
En remplaçant l'activité de l'oxygène par la pression partielle de dioxygène
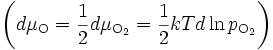 , on obtient
, on obtientEn intégrant cette équation sur l'épaisseur Δx de la couche, on obtient :

(19)
où
- N est le nombre de moles d'oxyde ;
- pIO2 et pIIO2 sont les pressions partielles d'oxygène aux interfaces oxyde/gaz et oxyde/métal ;
- kt est le coefficient d'oxydation parabolique.
Si l'on considère que la densité de l'oxyde est uniforme, l'épaisseur Δx de la couche est proportionnelle à la quantité d'oxyde N (Δx ∝ N) : le volume d'oxyde Vox s'écrit
où ρ est masse volumique de l'oxyde et MMnO2 est sa masse molaire. On a donc
et donc
donc en intégrant
puisqu'en début d'oxydation, la quantité d'oxyde est nulle (N = 0 à t = 0), soit
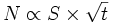
Comme la prise de masse est due à l'incorporation d'atomes d'oxygène, on a
- Δm = N⋅2⋅MO
où 2 est le nombre d'atomes d'oxygène par maille de MnO2, et MO est la masse molaire de l'oxygène (16 g·mol−1), donc
Ce sont donc bien des lois paraboliques.
Cas particuliers
Défauts prédominants et diffusion
Dans le cas général, on a deux défauts dominants : un défaut ionique (dans le sous-réseau d'oxygène ou de métal) et un défaut électronique (électron libre ou trou délectron).
En raison de la neutralité électrique, les défauts doivent avoir une charge de signe opposé, on a donc quatre possibilités :
- lacunes métalliques VMzc ' et des trous d'électron h•, l'oxyde est alors dit « en déficit métallique » ;
- ions métalliques interstitiels Mizc • et électrons libres e', l'oxyde est alors dit « en excès métallique » ;
- lacunes d'oxygène VO2 • et électrons libres e' ; l'oxyde est alors dit « en déficit d'oxygène » ;
- ions oxygène interstitiels Oi2 ' et trous d'électron h• ; l'oxyde est alors dit « en excès d'oxygène ».
Les électrons et trous d'électron étant beaucoup plus rapide que les ions, l'oxyde se comporte comme un semi-conducteur de type P dans les cas 1 et 4, et de type N dans les cas 2 et 3.
Dans certains cas, lorsque que l'oxyde est proche de la stœchiométrie, les deux défauts prédominants peuvent être ioniques et former des défauts de Frenkel VMzc ' + Mi2 • ou VO2 • + Oi2 ', ou des défauts de Schottky VMzc ' + VO2 •.
Du fait de la faible concentration en charges électroniques, le conducteur est de type ionique ; on parle « d'électrolyte solide ».
Dans une couche d'oxyde obtenue par réaction avec l'atmosphère, le type de défaut dominant peut varier en fonction de la profondeur puisque la pression partielle de dioxygène varie.
Les considérations ci-dessus ne sont valables que pour de faibles concentrations de défaut (jusqu'à environ 10−4 par maille, soit un défaut pour 10 000 mailles). Au delà, les défauts interagissent et ne peuvent plus être considérés comme indépendants. Pour des concentrations élevées (fraction supérieure à 10−2), les défaut forment des « associations (ou amas) de défauts » (defect clusters)[7]. Un ion interstitiel de charge positive (ion métallique ou impureté) ayant une charge relative opposée à la lacune métallique, la présence d'interstitiels a tendance à stabiliser les lacunes par attraction coulombienne (électrostatique) ; lorsque l'on a a interstitiels entourés de b lacunes, on parle de défaut (b : a)[8]. Un exemple simple est l'amas de défauts (4:1) dans le wüstite Fe1-zO (structure NaCl).
Oxyde ayant une conductivité électronique prédominante
On a tel ≈ 1 et tion << 1, soit
- σ⋅tion⋅tel ≃ σ⋅tion = σion
avec
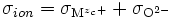 .
.
D'après (E9), on a donc
kt dépend donc des coefficients d'autodiffusion des anions et des cations dans l'oxyde (qui eux-mêmes dépendent de la pression de dioxygène dans l'oxyde) :
Or, les coefficients de diffusion DM et DO s'expriment en fonction des coefficients d'autodiffusion des défauts et de leur concentration, par exemple dans le cas d'un oxyde déficitaire en oxygène,
la concentration en défaut peut elle-même se déduire de la pression partielle de dioxygène. Ainsi, il est possible d'exprimer kt en fonction des paramètres fondamentaux du matériau et de l'environnement.
Oxyde ayant une conductivité ionique prédominante
Si l'oxyde a une conductivité ionique prédominante, alors la diffusion est limitée par le transport des électrons et des trous d'électron. On a tion ≈ 1 et tel << 1, soit
- σ⋅tion⋅tel ≃ σel
et donc
Les équilibres de défauts permettent de relier la concentration en électrons à celle des trous d'électrons en fonction de la pression partielle de dioxygène. Si par exemple on suppose que le défaut prédominant est le défaut de Schottky chargés et que les trous d'électron sont prédominants sur les électrons, on a
avec donc la constante d'équilibre
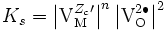 .
.
Par ailleurs, on a
avec la constante d'équilibre
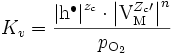 .
.
On peut ainsi exprimer le coefficient parabolique d'oxydation en fonction de Ks, Kv et de pO2 .
Annexes
Bibliographie
- E. Belorizky, W. Gorecki, Introduction à la mécanique statistique, éd. Presses universitaires de Grenoble, Grenoble (France), 1992.
- F. Gesmundo, Diffusion in Solids and High Temperature Oxidation of Metals, chapitre Transport processes during the oxidation of metals, pages 57–102, éd. Trans Tech Publications, 1992.
- P. Kofstad, High temperature corrosion, éd. Elsevier Applied Science, 1988.
- J. Philibert, A. Vignes, Y. Bréchet et P. Combrade, Métallurgie du minerai au matériau, éd. Dunod, Paris (France), 1998.
Notes et références
- ↑ Carl Wagner, physico-chimiste allemand (1901-1977) ; énonça sa loi parabolique en 1933, le premier à proposer une loi parabolique fut Tamman en 1920, avec un modèle plus simple.
- ↑ J. Philibert, A. Vignes, Y. Bréchet et P. Combrade, Métallurgie du minerai au matériau, éd. Dunod, Paris (France), 1998, p999
- ↑ P. Kofstad, High temperature corrosion, éd. Elsevier Applied Science, 1988, p163
- ↑ F. Gesmundo, Diffusion in Solids and High Temperature Oxidation of Metals, chapitre Transport processes during the oxidation of metals, pages 57–102, éd. Trans Tech Publications, 1992, p61
- ↑ Certains ouvrages notent ce flux de charge I, mais il y a alors un risque de confusion avec le courant électrique global.
- ↑ Dans sa forme intégrale, on retrouve évidemment U = RI où U est la différence de potentiel, R la résistance et I l'intensité du courant.
- ↑ F. Gesmundo, Diffusion in Solids and High Temperature Oxidation of Metals, chapitre Transport processes during the oxidation of metals, pages 57–102, éd. Trans Tech Publications, 1992, p73
- ↑ P. Kofstad, High temperature corrosion, éd. Elsevier Applied Science, 1988, p55
 Portail de la chimie
Portail de la chimie Portail de la physique
Portail de la physique
Catégorie : Corrosion
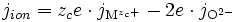
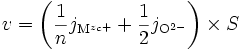
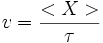
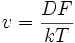
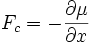
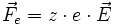
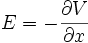
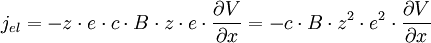
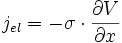
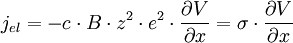
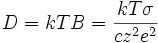
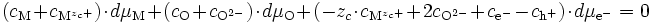
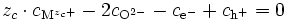
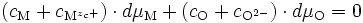
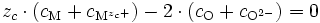
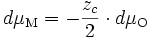
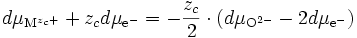
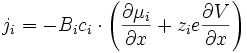
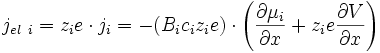
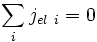
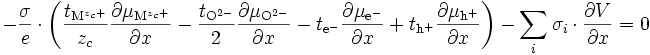
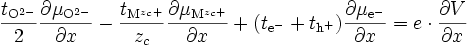
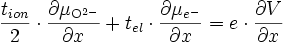
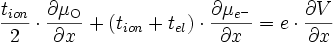
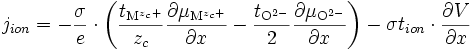
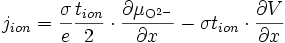
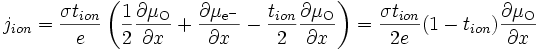
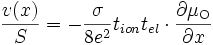
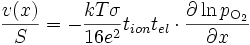
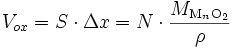
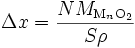
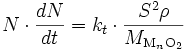
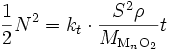
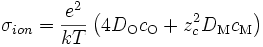
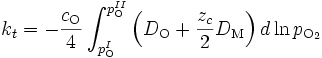
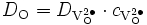
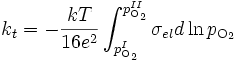
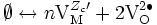
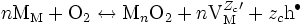
Wikimedia Foundation. 2010.