- Am1
-
AM1
AM1 ou Austin Model 1 est une méthode de calcul de chimie quantique développée par M. Dewar en 1985. Le modèle AM1 est basé sur une approche Hartree-Fock semi-empirique. À la différence d'une approche ab initio, où toutes les intégrales bi-électroniques sont calculées, on réalise dans une approche semi-empirique un certain nombre d'approximations de manière à réduire ce nombre d'intégrales et ainsi alléger le temps de calcul. Ces approximations sont les suivantes :
- Seuls les électrons de valence sont considérés explicitement dans les calculs (on considère que les électrons de cœur et le noyau forment un noyau effectif)
- Une base minimale est utilisée pour les électrons de valence
- La matrice recouvrement S est traitée selon l'approximation ZDO (Zero Differential Overlap)
L'approximation ZDO va annuler tous les produits de fonctions de base associées aux mêmes coordonnées ; on peut donc écrire pour les éléments de matrice de S :
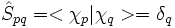
Il est évident que le fait d'introduire des approximations va avoir pour effet de s'écarter de la réalité. Les approximations sonr donc compensées en paramétrisant les intégrales restantes. Le nombre d'intégrales négligées ainsi que le type paramétrisation vont définissent les différentes méthodes semi-empiriques.
Le modèle AM1 fut développé dans le but d'améliorer le modèle MNDO (Modified Neglect of Differential Overlap) qui conduisait à des énergies de répulsion trop élevées. Le terme répulsif a alors été modifié par l'introduction de fonctions gaussiennes de forme mathématique :
![\hat V(A,B) = Z_A Z_B <s_As_A|s_Bs_B> [1 + F(A) + F(B)]](/pictures/frwiki/57/9e97edcc95ead6c946af2485338d90a9.png)
où
Les termes K, L et M sont des paramètres tandis que les indices i et j sont les nombres de fonctions gaussiennes impliquées dans les calculs. La valeur de ces indices varie typiquement de 2 à 4 selon l'atome considéré.
Références
- Dewar, M. J. S., Zoebisch, E. G., Healy, E. F. and Stewart J. J. P., Journal of the American Chemical Society, 107, 3902, (1985)
Voir aussi
 Portail de la physique
Portail de la physique Portail de la chimie
Portail de la chimie
Catégorie : Chimie quantique
![\hat F(A) = e^{-\alpha_AR_{AB}} + \sum_i e^{[L_{Ai}(R_{AB}-M_{Ai}^2]}](/pictures/frwiki/98/bbbb36f2f1fadb38148aa085ec5670da.png)
![\hat F(B) = e^{-\alpha_BR_{AB}} + \sum_j e^{[L_{Aj}(R_{AB}-M_{Aj}^2]}](/pictures/frwiki/54/62c7ef5a41e3ba867f4efd21a991fd5e.png)
Wikimedia Foundation. 2010.