- FRET (Physique)
-
Transfert d'énergie entre molécules fluorescentes
 Pour les articles homonymes, voir Fret (homonymie).
Pour les articles homonymes, voir Fret (homonymie).Introduction
Le transfert d'énergie entre molécules fluorescentes ou transfert d'énergie par resonance de type Förster (FRET, pour l'acronyme anglais "Förster resonance energy transfer"), bien qu’observé par Perrin au début du XXe siècle, est décrit pour la première fois par Förster en 1946. Les applications de cette approche à l’étude des interactions protéiques apparaîtront vers la fin du XXe siècle (Selvin, 2000).
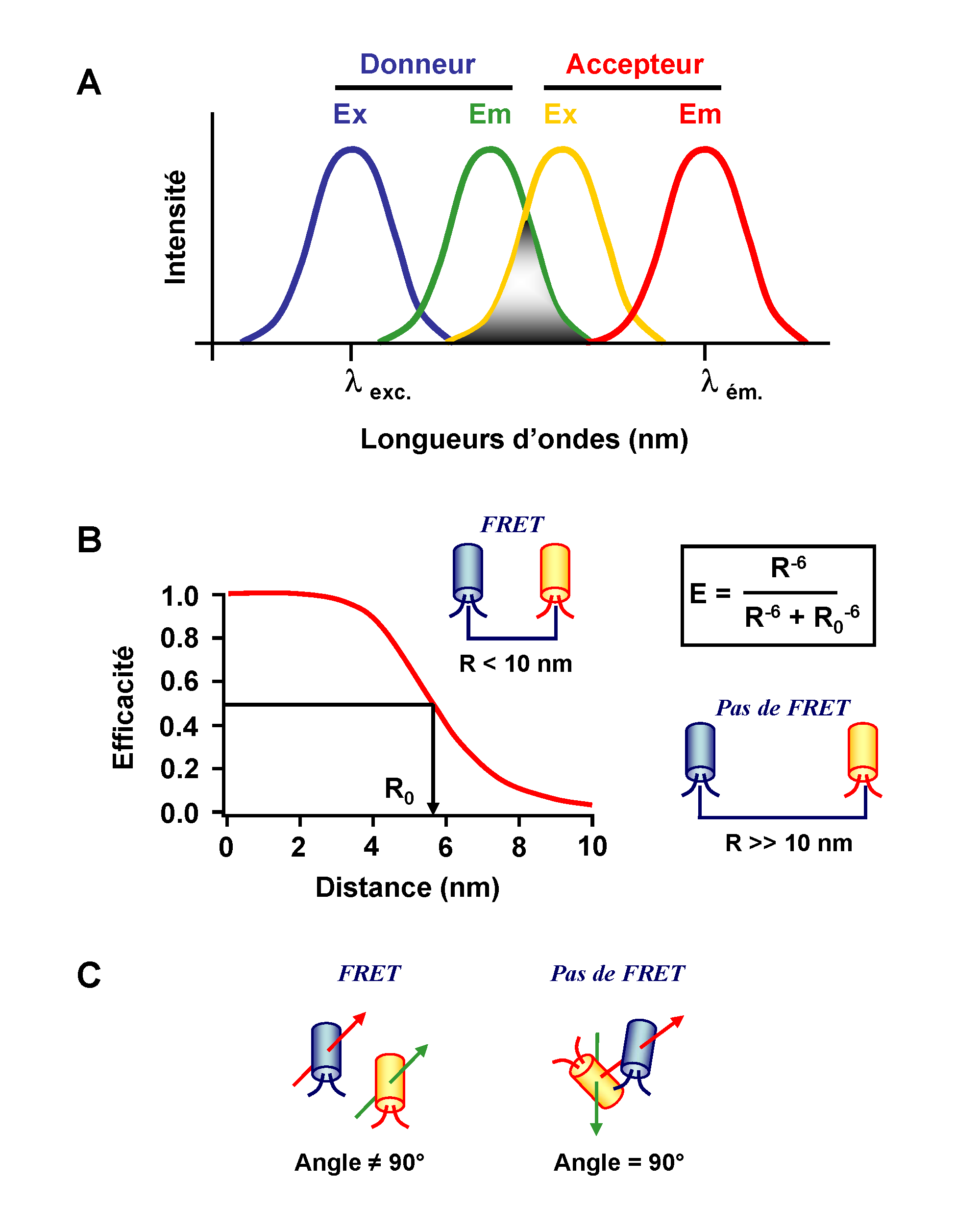
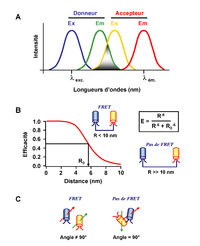 Figure 1. Conditions du FRET. A. Un FRET peut apparaître seulement si le spectre d’émission du donneur recouvre le spectre d’excitation de l’accepteur. Ce recouvrement est défini par une intégrale de recouvrement J. B. Un FRET est observé si la distance séparant les deux fluorophores est inférieure à 1.8 x le rayon de Förster (R0 ). Ce dernier définit la distance donneur – accepteur pour laquelle l’efficacité du transfert d’énergie est de 50%. C. Importance de l’orientation relative des dipôles du donneur et de l’accepteur pour la mise en place d’un transfert d’énergie.
Figure 1. Conditions du FRET. A. Un FRET peut apparaître seulement si le spectre d’émission du donneur recouvre le spectre d’excitation de l’accepteur. Ce recouvrement est défini par une intégrale de recouvrement J. B. Un FRET est observé si la distance séparant les deux fluorophores est inférieure à 1.8 x le rayon de Förster (R0 ). Ce dernier définit la distance donneur – accepteur pour laquelle l’efficacité du transfert d’énergie est de 50%. C. Importance de l’orientation relative des dipôles du donneur et de l’accepteur pour la mise en place d’un transfert d’énergie.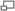
Conditions du transfert d’énergie
D’après la théorie de Förster, le FRET est défini comme un transfert d’énergie non radiatif (sans émission de lumière) résultant d’une interaction dipôle – dipôle entre deux molécules (donneur et accepteur d’énergie) (Stryer, 1978). Ce phénomène physique nécessite une compatibilité énergétique entre ces molécules. Cela signifie que le spectre d’émission du donneur doit recouvrir, au moins partiellement, le spectre d’absorption de l’accepteur (Figure 1.A). Ce recouvrement des spectres est défini par une intégrale appelée intégrale de recouvrement J(λ) :
où fD est l’intensité de la fluorescence émise par le donneur à une longueur d’onde donnée et εA le coefficient d’extinction molaire de l’accepteur. Le facteur J reflète donc la capacité d’une paire de fluorophores à émettre et absorber de l’énergie à la même longueur d’onde.
En accord avec la théorie de Förster, le FRET est un processus qui dépend de la distance séparant les deux molécules, donneur et accepteur, comme le montre la formule suivante :
où R est la distance effective qui sépare les deux molécules et R0 le rayon de Förster. Ce dernier correspond à la distance donneur - accepteur pour laquelle l’efficacité du transfert d’énergie est de 50% (Figure 1.B). Cette distance, qui dépend de la nature des fluorophores utilisés, est généralement comprise entre 1 et 10 nm (Stryer and Haugland, 1967). Au-delà de cette gamme, l’efficacité du transfert d’énergie chute très rapidement. L’expression mathématique pour le calcul de cette distance s’écrit :
où J est l’intégrale de recouvrement, n l’indice de réfraction du milieu (n − 4 est généralement compris entre 1/3 et 1/5), QD le rendement quantique du donneur en absence d’accepteur et κ2 le facteur d’orientation qui est fonction de l’orientation relative des dipôles du donneur et de l’accepteur (Figure 1.C). Même si la valeur de κ2 est théoriquement comprise entre 0 et 4, 2/3 est la valeur habituellement utilisée pour déterminer le R0. En effet, κ2 est assimilé à 2/3 lorsque le donneur et l’accepteur présentent un degré de liberté suffisant pour être aléatoirement orientés dans l’espace (Stryer, 1978). Cette condition est généralement satisfaite pour les fluorophores attachés à des biomolécules car ils peuvent avoir une certaine liberté de rotation (Dale et al., 1979).
Caractéristiques photophysiques d'un fluorophore
Une molécule capable d’émettre un signal de fluorescence est qualifiée de fluorophore. Différentes caractéristiques photophysiques permettent de le définir :
- les spectres d’excitation et d’émission. Ils représentent la signature de la structure énergétique du fluorophore. La différence de longueur d’onde séparant leur maximum s’appelle le déplacement de Stokes.
- le coefficient d’extinction molaire (ε). Il correspond à la capacité d’absorption par le fluorophore de l’énergie apportée par un photon à une longueur d’onde donnée.
- le rendement quantique (Φ). Il caractérise la capacité du fluorophore à re-émettre sous forme de lumière, l’énergie absorbée. Il est défini comme étant le rapport du nombre de photons émis sur le nombre de photons absorbés.
- la durée de vie de fluorescence (τ). Elle représente le temps de séjour moyen du fluorophore dans son état excité.
Voir aussi l'article sur la fluorescence.
Comment mettre en évidence le FRET ?
Le FRET mesure des intensités. Expérimentalement, ce signal peut être mesuré à l’aide d’un fluorimètre ou en microscopie. Dans un fluorimètre, le signal de FRET mesuré provient d'une population cellulaire disposée dans des puits de taille différente selon les microplaques utilisées. En revanche, les techniques microscopiques mesurent des évènements de transfert d’énergie à l’échelle subcellulaire. Quel que soit le type de détection choisi, la mesure d’un FRET entre deux fluorophores peut être effectuée de différentes manières.
La première consiste à quantifier les variations de l’intensité de fluorescence en mesurant la diminution de la fluorescence du donneur (Sokol et al., 1998), l’augmentation de celle de l’accepteur (Chan et al., 1979) ou en calculant un rapport que nous appelerons ratio (fluorescence d’émission de l’accepteur/fluorescence d’émission du donneur) (Miyawaki et al., 1997). Cette analyse est réalisable aussi bien en microplaque qu’en microscopie. La principale difficulté d’analyse de ces signaux vient du recouvrement pouvant exister entre les spectres d’excitation et d’émission des fluorophores utilisés. Ce manque de sélectivité spectrale est à l’origine d’un important bruit de fond avec comme conséquence une réduction de la sensibilité du test. Des couples de fluorophores tels que la fluorescéine/rhodamine ou les protéines CFP (Cyan fluorescent protein) / YFP (Yellow fluorescent protein) présentent ce type de limitation (Zheng et al., 2002). Cependant, le signal sur bruit du FRET peut être amélioré en s’affranchissant d’une partie des signaux parasites grâce à une lecture en temps résolu. Ceci est possible en TR-FRET (Time-Resolved Fluorescence Resonance Energy Transfer) grâce à l’utilisation de fluorophores à durée de vie longue tels que les chélates ou cryptates de terre rare. À ce jour, le TR-FRET est appliqué uniquement dans des formats microplaques.
D’autres méthodes permettent de déterminer indirectement l’existence d’un FRET par photoblanchiment (pbFRET). Le photoblanchiment est une méthode qui consiste à éteindre un fluorophore par une exposition prolongée à une source lumineuse. La mise en œuvre de cette approche est préférable en microscopie confocale car il est ainsi possible d’obtenir une bonne résolution spatiale pour éteindre directement les fluorophores dans leur environnement cellulaire. Dans le cas du FRET par photoblanchiment (pbFRET), le photoblanchiment du fluorophore donneur entraîne une diminution de son intensité de fluorescence qui est mesurée en présence ou non d’accepteur. Lorsque l’accepteur est en étroite proximité avec le donneur, un FRET apparaît et entre en compétition avec le processus de photoblanchiment. Cette compétition se traduit par une augmentation de la résistance du donneur au photoblanchiment dont les constantes de temps mesurées (en présence ou non d’accepteur) permettent de mettre en évidence un transfert d’énergie (Rocheville et al., 2000). Une autre alternative au pbFRET du donneur est le pbFRET de l’accepteur. Les émissions de fluorescence du donneur et de l’accepteur sont mesurées avant et après photoblanchiment de l’accepteur. L’augmentation de la fluorescence du donneur après destruction de l’accepteur prouve l’existence d’un FRET entre les deux molécules (Gregan et al., 2004; He et al., 2003). Un des désavantages du pbFRET est qu’une mesure répétée du même échantillon n’est pas envisageable en raison de la destruction des fluorophores par l’excitation continue.
L’utilisation de la durée de vie de fluorescence du donneur est aussi une approche adaptée à la mesure des évènements de FRET. Cette méthode consiste à mesurer le déclin de fluorescence du donneur au cours du temps. Cela peut être réalisé aussi bien sur des populations de cellules au format microplaque, que par des techniques microscopiques. La base physique de cette approche repose sur le fait que la durée de vie d’une molécule fluorescente dépend notamment de l’efficacité du processus de FRET. Par conséquent, plus le transfert d’énergie entre les deux molécules est efficace plus le déclin de fluorescence du donneur est rapide. Le FLIM (Fluorescence Lifetime Imaging Microscopy) est un exemple de technologie basée sur l’analyse de durée de vie (Bastiaens and Squire, 1999). Il est préférable avec ce type d’approche d’utiliser des molécules fluorescentes dont le déclin de fluorescence est monoexponentiel. La CFP par exemple est utilisée en FLIM en dépit de certains désavantages dus à son faible rendement quantique, son faible coefficient d’extinction molaire et surtout à son déclin de fluorescence qui présente deux exponentielles (Tramier et al., 2002). Ainsi, afin de faciliter l’analyse des durées de vie en FLIM des mutations de cette protéine ont permis de générer un variant nommé Cerulean plus lumineux que la CFP et présentant une décroissance monoexponentielle (Rizzo et al., 2004).
Utilisation de la GFP et de ses dérivés en FRET
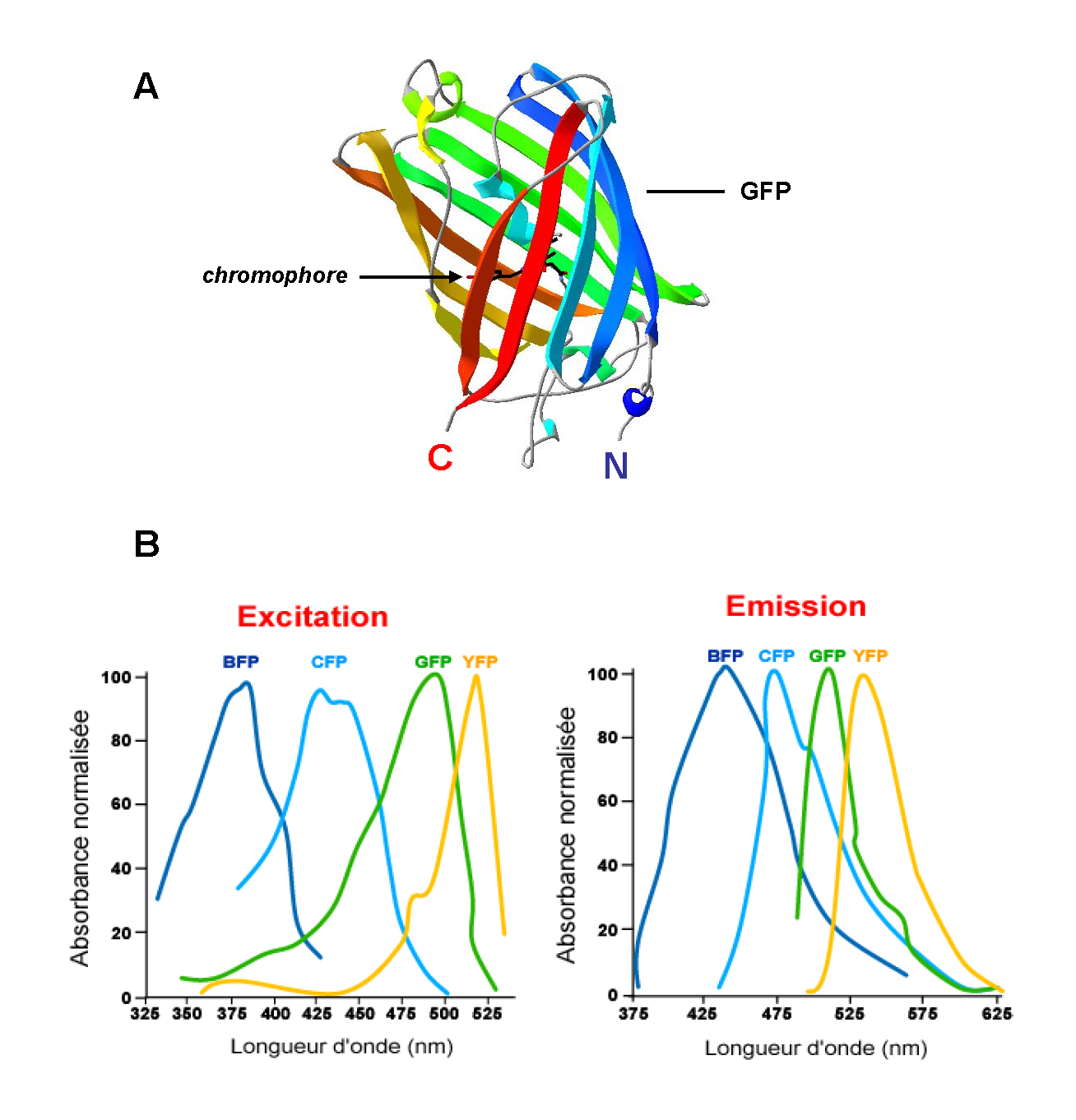
 Figure 2. Les protéines GFP. A. Structure cristallographique de la GFP (code pdb : 1EMB). B. Spectres d’excitation et d’émission des variants de la GFP.
Figure 2. Les protéines GFP. A. Structure cristallographique de la GFP (code pdb : 1EMB). B. Spectres d’excitation et d’émission des variants de la GFP.L’innovation probablement la plus importante dans le domaine des fluorophores fut marquée par la découverte d’une protéine fluorescente pouvant être directement encodée à l’intérieur de la cellule. Il s’agit de la GFP (Green Fluorescent Protein). L’exploitation de la GFP et la génération de variants de cette protéine ont permis le développement de technologies de FRET parfaitement adaptées à l’étude de la dynamique des interactions moléculaires dans l’espace et dans le temps à l’intérieur de cellules vivantes.
Initialement identifié chez la méduse Aequorea Victoria, le gène codant la GFP a été cloné en 1992 par Prasher et coll. Cristallisée pour la première fois en 1974, il faudra attendre 1996 pour obtenir la structure tridimensionnelle de la GFP par diffraction de rayons X (Ormo et al., 1996). Cette protéine de 27 kDa se présente sous forme d’un cylindre de 30 Å (Ångström) de diamètre et 40 Å de hauteur composé de onze feuillets β (9 à 13 résidus par feuillet) qui entourent une hélice α contenant le chromophore (Figure 2.A). Le chromophore résulte d'une réaction spontanée de cyclisation des chaînes latérales de trois acides aminés (Sérine65 – Tyrosine66 – Glycine67) à l’intérieur du tonneau. L’organisation des feuillets β en formant une véritable cage, confère au chromophore son propre environnement (Ward, 2006).
La photoactivation de cette protéine dont le pic d’absorption majoritaire est à 395 nm entraîne une réaction autocatalytique provoquant l’émission d’une lumière verte avec un pic d’émission à 508 nm. Différentes protéines mutantes de la GFP ont été produites. En changeant, entre autres, la structure des trois acides aminés formant le chromophore, il a été possible de modifier les longueurs d'onde d'absorption et d'émission de la GFP devenant alors, une BFP (Blue Fluorescent Protein), une YFP (Yellow Fluorescent Protein) ou de nombreux autres variants bien caractérisés (CFP, RFP…) (Figure 2.B). L’utilisation de ces différents mutants comme sondes moléculaires en font des outils de choix aussi bien dans l’étude de l’expression de gènes, le suivi de protéines et leur compartimentalisation cellulaire que dans l’analyse des interactions protéine – protéine par des approches de transfert d’énergie. Concernant ce dernier point, deux paires de fluorophores ont été largement utilisées. Ces molécules BFP/GFP et CFP/YFP dérivées de la GFP présentent en effet des propriétés photophysiques compatibles avec la mise en place d’un transfert d’énergie.
Le point fort de cette technologie réside dans l’encodage des protéines fluorescentes. Il est donc possible de suivre en microscopie la localisation cellulaire des protéines avec une bonne résolution spatiale. De plus, grâce à la forte intensité de fluorescence des molécules utilisées, les phénomènes de transfert d’énergie peuvent être visualisés directement dans la cellule permettant de définir à la fois la nature des protéines en interactions, la dynamique de ces phénomènes et leur localisation cellulaire.
Dans le domaine des récepteurs couplés aux protéines G (RCPG) cette approche a été beaucoup utilisée pour caractériser la dimérisation des récepteurs (association par paire des protéines formant un récepteur fonctionnel). La fusion des GFP au niveau de la région carboxy-terminale des récepteurs a permis d’analyser leur organisation sans pour autant altérer leur structure, fonction ou localisation dans les travaux publiés. Le principal inconvénient de cette approche pour l’étude de la dimérisation vient de la nécessité de passer par la microscopie confocale. En effet, en microscopie classique par épifluorescence ou dans un fluorimètre il n’est pas possible de séparer le signal provenant des récepteurs de surface de celui des récepteurs retenus dans les compartiments intracellulaires. De plus, la difficulté de miniaturisation de cette approche en microplaque ne permet pas d’étendre ce type d’analyse à du moyen ou haut débit.
Utilisation du BRET (Bioluminescence Resonance Energy Transfer)
Voir l'article sur le BRET
Le FRET en temps résolu
Généralités
La principale limite du FRET est liée au manque de sélectivité spectrale des fluorophores utilisés ainsi qu’à la difficulté de s’affranchir des signaux parasites (bruit de fond). Ceci a des conséquences sur la sensibilité des tests mis en œuvre. Ainsi, l’utilisation de traceurs présentant des propriétés originales de luminescence a permis de mettre au point des tests plus sensibles en améliorant la résolution spectrale et temporelle du signal de FRET. Ces molécules sont des complexes formés par l’association d’un chromophore (cryptand ou chélate) et d’un cation lanthanide faisant partie du groupe des terres rares (europium, terbium…). La principale caractéristique des lanthanides vient de leur durée de vie de luminescence relativement longue (de l’ordre de la milliseconde). La durée de vie de fluorescence de la plupart des fluorophores organiques et des protéines fluorescentes est de l’ordre de la nanoseconde, tout comme les fluorescences parasites (autofluorescence, diffraction de la lumière…). Grâce à cette propriété des ions lanthanides, les systèmes de détection de fluorescence en phase homogène et en temps résolu ont pu être développés. Ces systèmes reposent sur l’application d’un délai entre l’excitation de l’échantillon et la mesure du signal émis de manière à s’affranchir des fluorescences parasites à durée de vie courte. Cette résolution temporelle du signal permet ainsi d’améliorer le rapport entre le signal du traceur et le bruit de fond inhérent aux conditions du test sans qu’aucune étape de séparation des espèces ne soit nécessaire.
Les fluorophores
Le donneur
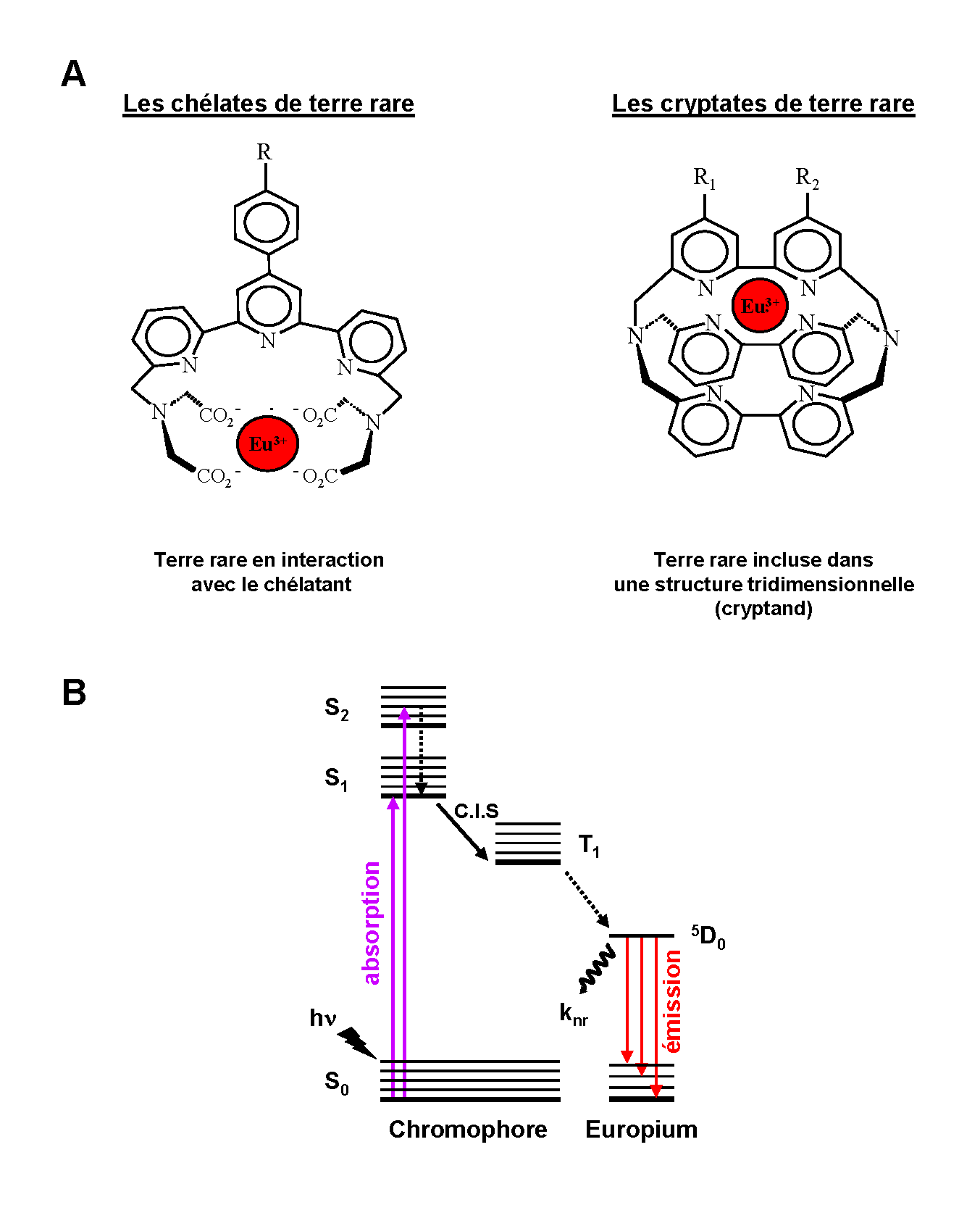
 Figure 3. Les chélates et cryptates de terre rare. A. Structure des chélates et cryptates de terre rare. B. Transfert d’énergie entre le chromophore (chélate ou cryptate) et l’ion europium. C.I.S = croisement intersystème.
Figure 3. Les chélates et cryptates de terre rare. A. Structure des chélates et cryptates de terre rare. B. Transfert d’énergie entre le chromophore (chélate ou cryptate) et l’ion europium. C.I.S = croisement intersystème.Au cours des années 1970, les complexes luminescents de lanthanide se sont révélés des candidats intéressants comme marqueurs dans le développement de systèmes d’analyse et de diagnostic. En effet, l’utilisation des chélates et cryptates de terre rare a permis de mettre au point des systèmes de détection en temps résolu caractérisés par une réduction du bruit de fond. Chacune de ces sondes est constituée d’un chromophore organique et d’une terre rare (principalement europium et terbium). La complexation de l’ion par le chélate est basée sur une interaction réversible alors que dans la structure du cryptate, le cryptand encage irréversiblement l’ion en le protégeant des interactions environnementales (extinction de fluorescence par les molécules d’eau…) (Figure 3.A). La présence de groupements réactifs au niveau du chromophore permet de greffer le complexe à des biomolécules (anticorps, antigènes…). Une propriété intéressante de ces chromophores est leur capacité à collecter l’énergie excitatrice (effet d’antenne) et à la transférer sur le cation lanthanide. L’antenne est nécessaire du fait de la faible capacité d’absorption de la lumière par les lanthanides qui rend leur excitation directe difficile (< fluorophores organiques conventionnels). Ainsi, l’excitation du complexe de lanthanide par une lumière incidente (laser, lampe flash…) contribue à peupler les niveaux vibrationnels singulets de haute énergie du chromophore (transition S0 → S1, S2) (Figure 3.B). Le retour vers l’état fondamental S0 est influencé par la présence du lanthanide qui favorise le croisement intersystème permettant de peupler les états triplets (T1) du chromophore. Finalement, le passage T1 → S0 induit un transfert d’énergie favorisant le remplissage des niveaux excités du lanthanide (5D0 pour l’europium et 5D4 pour le terbium) dont la composante de désexcitation par voie radiative est responsable de l’émission luminescente.
Le transfert d’énergie intramoléculaire existant au sein de ces complexes est responsable du grand déplacement de Stokes (de l’ordre de 200 – 250 nm) qui permet de s’affranchir de l’interférence due à la source d’excitation. Ce décalage est la conséquence de la séparation des fonctions absorption (par le chromophore) et émission (par la terre rare) au sein du complexe. Ainsi, les chromophores organiques (chélate, cryptate) absorbent majoritairement dans l’UV et la partie bleue du spectre visible alors que les terres rares réémettent dans le vert – rouge : 560 nm pour le terbium et 605 nm pour l’europium. La longue durée de vie de cette émission (µs → ms) est principalement la conséquence des transitions électroniques particulières qui interviennent au niveau des terres rares. Cette caractéristique permet d’ailleurs l’utilisation de ces molécules dans des applications en temps résolu.
Dans les milieux biologiques classiquement utilisés, le complexe formé par le chélate et le lanthanide peut être dissocié en raison d’une faible stabilité de l’interaction (compétition entre l’ion lanthanide et les ions Mn2+, Mg2+, Ca2+ ou chélation du lanthanide par de l’EDTA), ce qui constitue un inconvénient. Dans le cas des cryptates de terre rare l’inclusion de l’europium dans une cage tridimensionnelle formée par le cryptand empêche ces phénomènes de dissociation conférant au complexe une très haute stabilité.
L’accepteur
Transfert d’énergie : sélectivité temporelle et spectrale
La sélectivité temporelle
La sélectivité spectrale
Exemples d'application du FRET
Transfert d’énergie intermoléculaire
Les techniques de transfert d’énergie, FRET (variants de la GFP, anticorps marqués…) ou BRET, ont été beaucoup utilisées pour démontrer des interactions protéines – protéines aussi bien à l’intérieur de la cellule qu’à la surface cellulaire. Quelle que soit la méthode choisie, cette démonstration repose sur la détection d’un signal caractéristique reflétant la forte proximité, si ce n’est l’association, des protéines cibles. Dans le domaine des récepteurs couplés aux protéines G, ces approches ont révélé l’existence de récepteurs membranaires homo- ou hétérodimériques et ont contribué à mieux comprendre les mécanismes intervenant au cours de l’activation des protéines G. Elles ont aussi permis de développer, entre autres, des sondes sensibles à des seconds messagers comme par exemple l’AMPc (Zaccolo et al., 2000). En effet, l’AMPc en se fixant au niveau des sous-unités catalytiques et régulatrices de la PKA, entraîne une dissociation de ce complexe. Ainsi, en fusionnant les fluorophores au niveau des différentes sous-unités, il est possible de corréler directement la production d’AMPc intracellulaire à une diminution du signal de FRET.
Transfert d’énergie intramoléculaire (changements conformationnels)
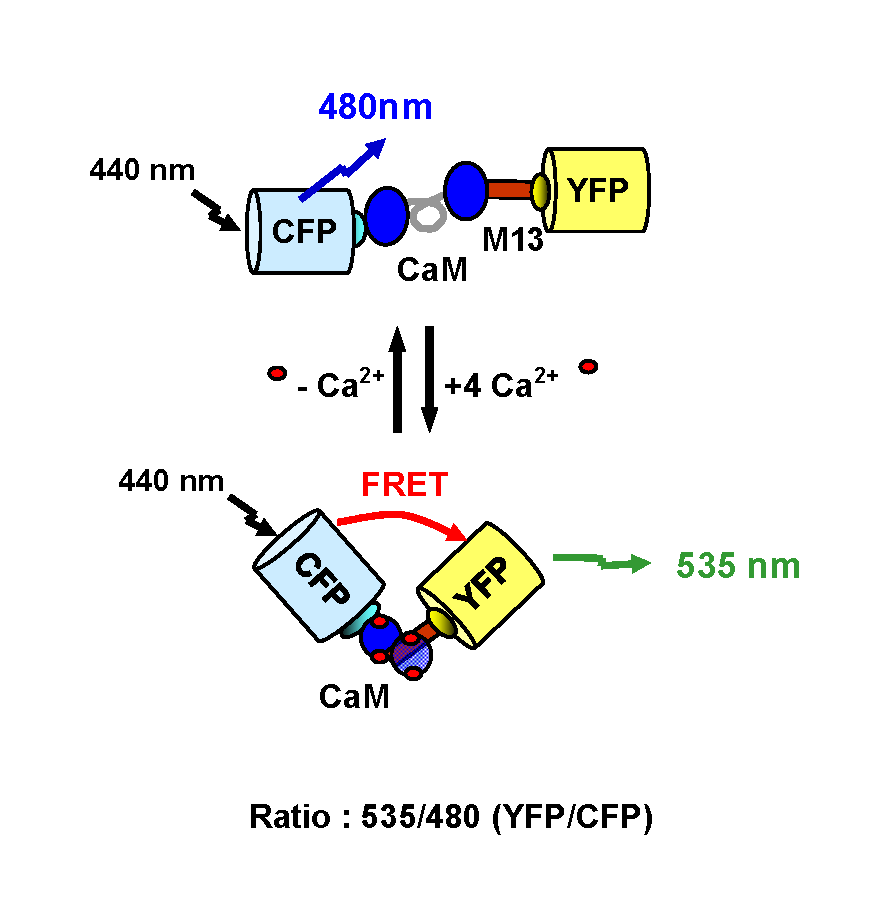
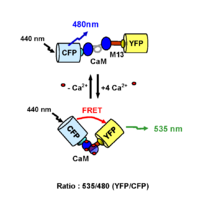 Figure. Transfert d’énergie intramoléculaire : sonde calcique (d’après Miyawaki et al., 1997).
Figure. Transfert d’énergie intramoléculaire : sonde calcique (d’après Miyawaki et al., 1997).Les techniques de transfert d’énergie sont des approches suffisamment sensibles pour mesurer des changements conformationnels intervenant au sein d’une protéine. Ceci est possible en incorporant les fluorophores donneur et accepteur à différentes positions de la protéine d’intérêt. Les points d’insertion sont choisis de manière à ce que les changements conformationnels, en modifiant la distance et/ou l’orientation des fluorophores, entraînent des modifications du transfert d’énergie (augmentation ou diminution).
Le transfert d’énergie intramoléculaire a permis d’analyser les changements conformationnels intervenant au cours des processus d’activation des récepteurs membranaires. Ainsi, Vilardaga et coll. ont analysé les réarrangements intracellulaires induits par la fixation de molécules agonistes et antagonistes au niveau des récepteurs béta-2A-adrénergiques et des récepteurs aux hormones parathyroïdiennes (Vilardaga et al., 2003). Le même type d’analyse a permis d’étudier les changements conformationnels du récepteur aux androgènes (Schaufele et al., 2005).
De nombreuses sondes intracellulaires ont aussi été développées sur ce principe. L’objectif de ces systèmes est de pouvoir révéler l’activation d’une voie de signalisation particulière en mesurant les variations de concentrations de différents seconds messagers. La première sonde fluorescente appelée caméléon, a été développée par Miyawaki et coll. afin de mesurer les modifications de la concentration calcique à l’intérieur de la cellule (Miyawaki et al., 1997) (Figure). Suite à la fixation de quatre ions calcium au niveau de son domaine calmoduline, la sonde subit des changements conformationnels provoquant un rapprochement des fluorophores. L’augmentation du signal de FRET à 510 (GFP) ou 535 nm (YFP) est dépendante de la production calcique intracellulaire. Il est ainsi possible de mesurer en temps réel, les variations de la concentration calcique à l’intérieur de la cellule. D’autres sondes ont aussi été développées pour mesurer notamment la concentration en GMPc (Nikolaev et al., 2006) ou pour détecter simultanément l’activité de la PKA et de la PKC (Brumbaugh et al., 2006).
Voir aussi
Bibliographie
- Lakowitz J.R.; Principles of fluorescence spectroscopy, second edition; Kluwer Academic/Plenum publisher.
 Portail de la chimie
Portail de la chimie Portail de la physique
Portail de la physique
Catégories : Optique | Chimie physique
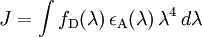
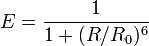
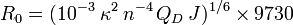
Wikimedia Foundation. 2010.