- Sarcoïdose
-
Sarcoïdose
Classification et ressources externes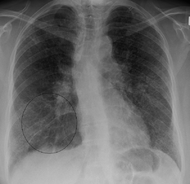
Radiographie du thorax montrant un infiltrat interstitiel. CIM-10 D86 CIM-9 135 OMIM 181000 DiseasesDB 11797 MedlinePlus 000076 eMedicine med/2063 MeSH D012507 La sarcoïdose ou maladie de Besnier-Boeck-Schaumann (communément dénommée BBS) ou lymphogranulomatose bénigne est une maladie inflammatoire systémique de cause inconnue, qui atteint préférentiellement les poumons, mais peut atteindre n'importe quels autres organes.
Généralement sans gravité, elle guérit spontanément sauf chez 20 % des malades, chez lesquels elle provoque des complications respiratoires menaçantes, ce qui justifie un diagnostic précoce et un suivi régulier.
Il n'existe pas à ce jour de traitement spécifique et les indications pour débuter un traitement sont rares.Sommaire
Épidémiologie
La sarcoïdose touche des hommes ou femmes de tout âge (généralement entre 30 et 50 ans) et de toute origine ethnique.
Chez les caucasiens, on note une égale fréquence de survenue chez les hommes et chez les femmes. La population noire (Africains et Antillais) est plus souvent touchée[1] avec une prédominance féminine. Les formes de sarcoïdose dans ces populations sont en outre plus graves[2].Son incidence varie de par le monde, avec, par exemple, pour l'Europe, un gradient nord-sud (640 pour 100 000 habitants en Suède contre 0,4 pour 100 000 habitants en Espagne), un gradient sud-nord aux USA (proportion de noirs-américains plus importante au sud). En France, la prévalence est de 10 pour 100 000 habitants.
Dans la plupart des cas, la sarcoïdose affecte l'interstitium pulmonaire et les ganglions médiastinaux.
Causes
Elles restent inconnues. Le risque de développer la maladie semble augmenter en cas d'exposition à des insecticides ou une vie en milieu agricole[3]. Il existe probablement un facteur génétique : il existe des formes familiales[4], chez des jumeaux[5] et un risque multiplié par cinq si un membre proche de la famille est atteint[6]. Une mutation sur le gène BTNL2 codant pour une immunoglobuline pourrait intervenir dans le développement de la maladie[7].
Physiopathologie
Le mécanisme de la survenue de la maladie reste flou. l'une des hypothèses fait intervenir une immunité à médiation cellulaire contre un antigène, pour l'instant inconnu, sur un terrain génétique prédisposé[8]. L'un des possibles antigènes serait une mycobactérie, une catalase-peroxydase mycobactérienne étant retrouvée dans la moitié des cas[9].
Les mécanismes immunologiques responsables de la sarcoïdose sont connus. La chronologie de l’inflammation sarcoïdosique a été particulièrement bien étudiée au niveau du poumon. Elle se caractérise par des désordres immunologiques, qui expliquent ses répercussions sur l’organisme. On distingue trois phases différentes qui se succèdent : l’alvéolite lymphocytaire et macrophagique, la phase granulomateuse, et finalement la fibrose (facultative).
- l’alvéolite lymphocytaire et macrophagique : Le système immunitaire produit une réponse très forte à un antigène inconnu, ce qui entraîne :
- une accumulation de lymphocytes CD8+ activés qui se concentrent dans les alvéoles du poumon (provoquant une alvéolite) Ces lymphocytes sécrètent des molécules pro-inflammatoires (l’interleukine 2 et un facteur chimiotactique des monocytes MCP-1. ce sont des médiateurs de l'inflammation : Il-2 par exemple) ;
- ils entraînent aussi une activation des lymphocytes B, qui sécrètent en réponse des anticorps (les gammaglobulines) ;
- des macrophages activés sont attirés par l'activation des lymphocytes CD4.
- la phase granulomateuse :
- les macrophages activés s'accumulent aussi dans le poumon. Ils secrètent notamment l'enzyme de conversion de l'angiotensine et forment des granulomes tuberculoïdes ;
- une anergie tuberculinique est fréquente (absence de réaction du corps au test de la tuberculose malgré une vaccination par le BCG), liée à une lymphopénie (baisse du nombre de lymphocytes « normaux », mais il n'y a pas d'immunodépression).
- la fibrose :
- à terme, les granulomes entraînent une fibrose pulmonaire, principale complication pulmonaire de la sarcoïdose.
Signes cliniques
L' atteinte des poumons est quasi constante : il y a 90 % de chance que les poumons soient atteints. Cependant les symptômes tels que des douleurs à la poitrine, essoufflements ou une toux sèche ne se manifestent que chez 33 % des personnes concernées.
Chez 33 % des personnes atteintes on observe une inflammation des ganglions.
La peau est atteinte dans un tiers des cas[8], avec apparition de petites boules de couleur rouge, très douloureuses (érythème noueux), avec - éventuellement - des taches ou plaques d'aspect peu spécifique. La reconnaissance des lésions cutanées est importante car la biopsie de ces dernières est facile et amène le diagnostic.
L’œil est touché dans 10 à 25 % des cas, avec rougeur, vision floue, démangeaisons. Ces signes correspondent à une uvéite, typiquement antérieure et chronique. l'examen à la lampe à fente montre des précipités bilatéraux, des adhérences (synéchies, des nodules de l'iris[10]. Une conjonctivite est souvent associée.
Les douleurs articulaires sont fréquentes. Elles sont dues à des arthrites (inflammation des articulations). Une augmentation du taux de calcium sanguin (hypercalcémie) n’est présente que dans 5% des cas. Cela peut entrainer une fatigue importante et des nausées.
L’atteinte du cœur se voit dans 4 cas sur 10[11]. Elle peut être silencieuse ou se manifester par des malaises, des signes d'insuffisance cardiaque. Rarement, une mort subite peut survenir. L'échocardiographie et l'IRM cardiaque peuvent montrer des anomalies non spécifiques.
L’atteinte du foie représente 20 % des cas. Cependant il n’y a pas souvent de symptômes (10 %).
Les signes observés, entre autres, dans le cas d'une neurosarcoïdose (atteinte cérébrale / système nerveux), peuvent être des vertiges rotatoires, perte d'équilibre, ceci est une forme des plus complexes et graves de la sarcoïdose. Cette atteinte n'est décrite que dans moins d'un cas sur dix[12]. Un syndrome dépressif peut se voir dans un peu moins d'un cas sur deux[8].
On peut observer une insuffisance rénale, des formations de calculs dans les urines ou une inflammation du nez et des sinus.
Dans sa forme typique, le syndrome de Löfgren associant érythème noueux, polyadénopathie et douleurs articulaires diffuses, suffit à faire poser le diagnostic sans que cela nécessite d'autres examens complémentaires.
Diagnostic
Le diagnostic n'est guère évident et peut-être retardé, particulièrement dans les formes uniquement pulmonaires où le délai peut dépasser six mois[13].
L'établissement du diagnostic nécessite le recueil d'éléments de plusieurs ordres :
- cliniques : signes présentés par le patient (souvent aucun) ;
- radiologiques, où on distingue quatre stades :
-
- stade 1 : adénopathies médiastinales bilatérales et symétriques ;
- stade 2 : adénopathies médiastinales et infiltrat/micronodules interstitiels ;
- stade 3 : infiltrat/micronodules interstitiels sans adénopathies médiastinales ;
- stade 4 : fibrose ;
- biologiques : absence de syndrome inflammatoire (sauf cas du syndrome de Löfgren), lymphopénie (diminution du nombre des lymphocytes), hypercalcémie (élévation de la concentration sérique de calcium libre), élévation de la concentration sérique de l'enzyme de conversion de l'angiotensine ;
- histologiques : la biopsie d'un granulome est nécessaire pour affirmer le diagnostic, sauf s'il existe un syndrome de Löfgren qui suffit à poser le diagnostic. On cherche le granulome le plus facilement accessible (peau, glande salivaire, adénopathie, bronches) dont l’étude microscopique objective un granulome sans nécrose caséeuse. la tomographie par émission de positons permet de détecter des atteintes non évidentes[14] et d'orienter une biopsie.
- le lavage broncho-alvéolaire se caractérise par une hyperlymphocytose à lymphocytes CD4 dans les alvéoles pulmonaires (accumulation des lymphocytes dans les poumons.)
Le diagnostic de la sarcoïdose est anatomo-pathologique, on doit retrouver un granulome épithélioïde et gigantocellulaire sans nécrose caséeuse sur une biopsie de l'organe atteint (biopsie d'adénopathies le plus souvent). La biopsie du granulome qui permet le diagnostic de certitude n'est indiquée que dans les formes modérées à sévères qui nécessiteront un traitement.
Examens complémentaires
En cas de diagnostic de sarcoïdose, différents examens sont pratiqués pour déterminer quels sont les organes touchés par la maladie. Ces examens permettent de détecter les lésions de façon précoce et d’assurer ainsi un suivi médical adapté. On distingue deux grandes familles d’examens : les examens mettant en évidence une atteinte intra pulmonaire et ceux mettant en évidence une atteinte extra pulmonaire. Les atteintes intra pulmonaires peuvent être diagnostiqué grâce à :
- Les épreuves fonctionnelles respiratoires (EFR) permettent d’apprécier la sévérité, le retentissement de l’atteinte pulmonaire et l'évolution. Ce test est indolore et dure une trentaine de minutes. Typiquement, on retrouve :
- une spirométrie normale ou un syndrome restrictif ;
- une analyse des gaz du sang normaux, une désaturation à l'effort, parfois une hypoxémie et une hypocapnie dans les formes sévères ;
- un abaissement de la DLCO et de la DLCO rapportée au volume alvéolaire (KCO)
- La fibroscopie qui permettra un lavage broncho-alvéolaire et une biopsie des lésions granulomateuses n'est indiquée que dans les formes suffisamment sévères pour justifier un traitement.
- La tomodensitométrie (ou scanner) est indiquée pour surveiller les formes sévères et diagnostiquer des lésions de fibrose. Cet examen permet d’évaluer plus précisément l’état des poumons et de visualiser des lésions invisibles en radiographie standard.
Les atteintes extra pulmonaires imposent un traitement urgent et efficace. Afin de les diagnostiquer, on réalise les examens suivants :
- un examen ophtalmologique ;
- des examens cardiaques, notamment la scintigraphie au Thallium, l’IRM et l’électrocardiogramme ;
- des explorations neurologiques : dans ces cas, l'apport du scanner et de l'IRM sont primordiaux. ;
- un bilan rénal.
Diagnostic différentiel
Le diagnostic différentiel repose[15],[16] sur deux modalités :
- la présentation radioclinique,
- la présence de lésions granulomateuses,
Avec granulome Sans granulome Dans tous les cas Suspecter
la tuberculose...
une vascularite[17],
et (plus rarement) ;
l'histoplasmose (Cf. contexte épidémiologique),
un traitement par interféron (par exemple pour hépatite C[18],[19]),
la bérylliose (exposition professionnelle en général, avec intoxication au béryllium ; tableau n° 33 des maladies professionnelles) ; La présence de béryllium dans l'organisme ou une réponse immunitaire à ce métal indiquant qu'il y a eu sensibilisation du patient, d'autant que la plupart des patients victimes de cette dernière ne savent pas qu'ils ont été en contact avec du béryllium[20]. Un test, des analyses et l'interrogation du patient sur un possible contact avec du béryllium peuvent orienter le diagnostic.Adénopathies médiastinales primo-infection de Tuberculose (rarement) si adénopathies unilatérales, lymphomes hodgkinien ou non (surtout en cas d'adénopathies antérieures) métastases des cancers solides au stade clinique (Cancer du poumon en général) Atteinte pulmonaire Pneumopathie d'hypersensibilité (médicamenteuse ou interstitielle idiopathique ; UIP, NSIP, AIP, BOOP, DIP, LIP) Lymphangite carcinomateuse (surtout secondaire à un cancer médullaire de la thyroïde, histiocytose X, fibrose intersticielle diffuse primitive, amyloïdose pulmonaire diffuse Atteinte extrathoracique Lèpre,
Syphilis,
Lymphogranulomatose d'inoculation, granulomatose médicamenteuse,
Maladie de Crohn,
Cancer broncho-pulmonaire,
Maladie de Whipple...Évolutions
La sarcoïdose est une maladie qui disparaît dans 50 % des cas en moins de trois ans[21]. Le syndrome de Löfgren en particulier, guérit seul dans 90 % des cas. La mortalité associée à la maladie est comprise entre 0,5 % et 5%[22]. Des complications sont possibles, mais très rares : atteinte cardiaque, méningite, fibrose respiratoire, insuffisance respiratoire chronique. l'apparition d'une hypertension artérielle pulmonaire est péjorative[23]. En dehors des cas sévères, aucun traitement n'est nécessaire (sinon, on propose une corticothérapie)
Traitement
La sarcoïdose peut guérir spontanément dans la majorité des cas. Pour cela, il faut distinguer entre les deux formes de la maladies : asymptomatique et symptomatique. Dans le premier cas, il n’est pas nécessaire de suivre un traitement quelconque. Un suivi clinique et biologique pendant une durée de six mois serait largement suffisant.
Les formes symptomatiques nécessitent un traitement médicamenteux notamment dans les formes de plus mauvais pronostic.
La prise en charge de la sarcoïdose a fait l'objet de plusieurs recommandation publiées par des sociétés savantes nationales ou internationales, dont celles de l' American Thoracic Society, datant de 1999[24], et celles, datant de 2008 de la British Thoracic Society[25].
Corticothérapie
Le traitement le plus utilisé est la corticothérapie (traitement par corticoïdes), que ce soit localement (corticoïde sous forme inhalée, locale pour les uvéites antérieures ou injectable pour les uvéites postérieures) ou par voie générale.
Les indications formelles à la corticothérapie orale sont les localisations entrainant un risque vital ou fonctionnel majeur (cœur, système nerveux central, œil, muscle, hypercalcémie persistante) ou les localisations cutanées importantes (préjudice esthétique). Dans les formes seulement pulmonaires, seuls les patients symptomatiques présentant des anomalies radiologiques et fonctionnelles significatives doivent être traités d'emblée.
Le traitement corticoïde est prolongé, d'au moins 2 ans, à la dose initiale de 0.5 mg/kg/jour de prednisolone (parfois jusqu'à 1 mg/kg/jour). Les doses seront ensuite très lentement dégressives, avec de possibles rechutes lorsque les doses deviennent inférieures à 0.15 mg par jour et à l'arrêt du traitement. Dans certains traitement, la corticothérapie peut être instaurée avec en même temps le methotrexate en injection sous cutanée hebdomadaire ou sous forme de comprimés.
L'efficacité de la corticothérapie est meilleure si le traitement est instauré avant l'apparition de lésions irréversibles.
Alternatives à la corticothérapie
- Les immunosuppresseurs : Pentoxyfilline, Thalidomide, Méthotrexate, Azathioprine (Imurel®), Ciclosporine A. Le méthotrexate pourrait, en particulier, diminuer les besoins en corticoïdes[26]. La cyclosporine s'avère non efficace dans cette indication[27].
- Les antipaludéens de synthèse (surtout pour les formes exclusivement cutanées)
- Les anticorps dirigé contre le TNF (tumor necrotizing factor[28] : infliximab, etanercept ou adalimumab. Les résultats sont, pour l'instant, décevants[29],[30].
D'autres molécules ont été testés : le Pentoxifylline pourrait diminuer les besoins en corticoïdes[31].
Les antioxydants n'ont aucun intérêt démontré dans la prise en charge de la sarcoïdose.
Historique
Jonathan Hutchinson (1828-1913) est le premier à avoir décrit la sarcoïdose en 1877[32].
Le dermatologue français Ernest Henri Besnier (1831-1909) décrivit en 1889 une lésion de la peau symétrique des extrémités.
Le dermatologue suédois Cæsar Peter Møller Boeck (1845-1917) mentionna en 1899 les lésions histologiques de la peau[33] et posa déjà alors le soupçon d’une maladie systémique. C’est pourquoi ces lésions sont appelés depuis lors comme étant la sarcoïdose de Boeck.
L’ophtalmologue danois Christian Frederick Heerfordt (1871-1953) décrit une infection fiévreuse de la conjonctive et à cause des analyses du laboratoire la classe comme étant due aux oreillons.
En 1924, le dermatologue suédois Jörgen Nilsen Schaumann (1879-1953) confirme la découverte de Boeck et qu’il s’agit d’une maladie systémique de plusieurs organes et dénomma la sarcoïdose « lymphogranulomatosis benigna », pour la distinguer du lymphome de Hodgkin.
Le Suédois Sven Halvar Löfgren (1910–1978) décrit en 1953 la forme aiguë, les « Trias », avec érythème noueux, arthrite et adénopathies ganglionnaires bi-hilaires
Associations d'entraide
La sarcoïdose étant déjà connue depuis plus de cent ans et concernant une partie non négligeable de la population d’âge moyen, des organisations internationales se sont constituées comme pour d’autres maladies, ainsi les personnes concernées ont pu s’organiser et créer des cercles d’entraide. Ceux-ci travaillent souvent ensemble et se sont donnés pratiquement les mêmes buts.
Un des buts principaux est l’information sur la sarcoïdose des personnes atteintes et de leur entourage. Il leur est le plus souvent très difficile de trouver des informations destinées au grand public. Vu les symptômes très variés et la nécessité de diagnostics différentiels et des connaissances relativement infimes de beaucoup de médecins en ce qui concerne cette maladie, ils doivent en être informés également d’une façon plus vaste. Un autre but est la sensibilisation du grand public pour les problèmes des malades, qui à cause de leurs symptômes diffus, sont placés dans la catégorie des « simulant ».
Aussi une meilleure qualité des soins et la prise en charge des patients atteints de sarcoïdose sont un des buts principaux des organisations. Souvent sont de même demandés une meilleure structure pour les soins et des services ambulatoires spécialisés, ainsi qu’une recherche accrue des causes et des moyens thérapeutiques et de soulagement. La sensibilisation de la sphère politique, ainsi que des responsables de la santé et du social, joue aussi un rôle primordial. Beaucoup d’organisations d’entraide pour la sarcoïdose sont actives pour promouvoir la recherche concernant cette pathologie.
Références
- Rybicki BA, Major M, Popovich J Jr, Maliarik MJ, Iannuzzi MC, Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization, Am J Epidemiol, 1997;145:234–241
- La sarcoïdose par le professeur B. Wallaert, université de Lille. Présentation du Dr I. Ghozlani.
- Newman LS, Rose CS, Bresnitz EA et Als. ACCESS Research Group. A Case Control Etiologic Study of Sarcoidosis: environmental and occupational risk factors, Am J Respir Crit Care Med, 2004;170:1324–1330
- Rybicki BA, Iannuzzi MC, Frederick MM, Thompson BW, Rossman MD, Bresnitz EA et als. Familial aggregation of sarcoidosis. A case-control etiologic study of sarcoidosis, Am J Respir Crit Care Med, 2001;164:2085-91
- Sverrild A, Backer V, Kyvik KO, Kaprio J, Milman N, Svendsen CB et als. Heredity in sarcoidosis: a registry-based twin study, Thorax, 2008;63:894-6
- Iannuzzi MC, Genetics of sarcoidosis, Semin Respir Crit Care Med, 2007;28:15-21
- Valentonyte R, Hampe J, Huse K et Als. Sarcoidosis is associated with a truncating splice site mutation in BTNL2, Nat Genet, 2005;37:357–364
- Iannuzzi MC, Fontana JR, Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics, JAMA, 2011;305:391-399
- Song Z, Marzilli L, Greenlee BM et Als. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis, J Exp Med, 2005;201:755–767
- Bonfioli AA, Orefice F, Sarcoidosis, Semin Ophthalmol, 2005;20:177–182
- Mehta D, Lubitz SA, Frankel Z et al. Cardiac involvement in patients with sarcoidosis: diagnostic and prognostic value of outpatient testing, Chest, 2008;133:1426–1435
- Lower EE, Weiss KL, Neurosarcoidosis, Clin Chest Med, 2008;29:475
- Judson MA, Thompson BW, Rabin DL et Als. ACCESS Research Group. The diagnostic pathway to sarcoidosis, Chest, 2003;123:406–412
- Teirstein AS, Machac J, Almeida O et Als. Results of 188 whole-body fluorodeoxyglucose positron emission tomography scans in 137 patients with sarcoidosis, Chest, 2007;132:1949–1953
- Diagnostic différentiel de la sarcoïdose ; Medinfo, consulté 2011-08-07
- Sarcoïdose dans la base de données RESPIR (e-Revue mensuelle consacrée aux maladies respiratoires)
- Fernandes SR, Singsen BH, Hoffman GS. Sarcoidosis and systemic vasculitis. Semin Arthritis Rheum 2000;30:33-46
- Hoffmann RM, Jung MC, Motz R, Gössl C, Emslander HP, Zachoval R, Pape GR. Sarcoidosis associated with interferon-alpha therapy for chronic hepatitis C. J Hepatol 1998;28 :1058-63
- Cacoub P, Sbaï A, Francès C, Génesti C, Hausfater P, Piette JC. Systemic sarcoidosis during interferon-alpha therapy for chronic hepatitis C virus infection. Gastroenterol Clin Biol 2000;24:364-6
- Dossier INRS : Le béryllium, métal discret mais dangereux (mis en ligne Avril 2009)
- "La sarcoïdose, maladie de Besnier - Boeck - Schaumann"
- "Sarcoïdose"
- Shorr AF, Helman DL, Davies DB, Nathan SD, Pulmonary hypertension in advanced sarcoidosis: epidemiology and clinical characteristics, Eur Respir J, 2005;25:783–788
- American Thoracic Society, Statement on sarcoidosis, Am J Resp Crit Care Med, 1999;160:736-55
- Bradley B, Branley HM, Egan JJ, Greaves MS, Hansell DM, Harrison NK et als. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society, Thorax, 2008;63(suppl 5):v1-58
- aughman RP, Winget DB, Lower EE, Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial, Sarcoidosis Vasc Diffuse Lung Dis, 2000;17:60–66
- Wyser CP, van Schalkwyk EM, Alheit B, Bardin PG, Joubert JR, Treatment of progressive pulmonary sarcoidosis with cyclosporin A: a randomized controlled trial, Am J Respir Crit Care Med, 1997;156:1371–1376
- Baughman RP, Lower EE, Drent M, Inhibitors of tumor necrosis factor (TNF) in sarcoidosis: who, what, and how to use them, Sarcoidosis Vasc Diffuse Lung Dis, 2008;25:76-89
- Utz JP, Limper AH, Kalra S et Als. Etanercept for the treatment of stage II and III progressive pulmonary sarcoidosis, Chest, 2003;124:177–185
- Baughman RP, Drent M, Kavuru M et Als. Sarcoidosis Investigators. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement, Am J Respir Crit Care Med, 2006;174:795–802
- Park MK, Fontana JR Jr, Babaali H et Als. Steroid-sparing effects of pentoxifylline in pulmonary sarcoidosis, Sarcoidosis Vasc Diffuse Lung Dis, 2009;26:121–131
- Hutchinson J, Anomalous diseases of skin and fingers: case of livid papillary psoriasis? In: Illustrations of clinical surgery. London: J and A Churchill, 1877:42-3
- Boeck C, Multiple benign sarcoid of the skin, J Cutan Genitourin Did, 1899;17543–550
Liens externes
- Europe
- European European Association of Patients Organizations of Sarcoidosis and other Granulomatous Disorders
- (Classé par ordre alphabétique) :
-
-
- Allemagne : DSV - Deutsche Sarkoidose Vereinigung
- Belgique : Vereniging voor sarcoïdosepatiënten (néerlandais)
- France : RISMS-Réseau International de Soutien des Malades de la Sarcoïdose
- France : Sarcoidose Infos, Site francophone sur la sarcoïdose
- Grèce : sarcoidosis.gr
- Italie : Site de l'Association italienne
- Norvège : Norsk Sarkoidose Forening
-
- **Pays-Bas : Sarcoidose Belangenvereniging Nederland SBN
- Autres pays :
Catégories :- Maladie du système immunitaire
- Maladie de la circulation pulmonaire
- l’alvéolite lymphocytaire et macrophagique : Le système immunitaire produit une réponse très forte à un antigène inconnu, ce qui entraîne :
Wikimedia Foundation. 2010.